Acid choice flips enantioselectivity
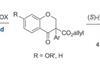
Changing proton source in asymmetric decarboxylation unexpectedly delivers opposite configuration
Changing proton source in asymmetric decarboxylation unexpectedly delivers opposite configuration