Algorithm designs proteins from scratch that can bind drugs and small molecules
By  Tim Wogan2024-04-12T08:32:00
Tim Wogan2024-04-12T08:32:00
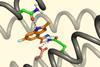
Strategy could stop an overdose or produce an antidote to a poison
A new algorithm based on physical principles can effectively design proteins that bind cancer drugs to potentially head off an overdose or allergic reaction. The researchers believe that the algorithm could potentially be more effective than deep learning approaches for designing proteins with unfamiliar requirements. This could make it useful for producing antidotes or delivery vehicles for drugs.1
Protein structure prediction has advanced hugely in recent years because of deep learning algorithms such as Deepmind’s AlphaFold and the Institute for Protein Design’s RoseTTAFold. These scour known structures in the Protein Data Bank and learn to predict how unknown structures will fold. They work purely using probability, however: energy does not appear explicitly in their calculations. ‘Deep learning could be learning physical principles,’ says Nicholas Polizzi of Harvard Medical School in Massachusetts, US, ‘and the way to test that is to design things with it that are totally outside the distribution of the training data. If it’s successful, you know it really did learn something and is able to generalise. So far, I haven’t really seen that with deep learning in protein design.’