
Observations of the oxygen–ruthenium bond could help improve catalyst design
Sometimes textbook concepts can be surprisingly challenging to demonstrate directly. The idea that overlap of electron orbitals gives rise to bonding and anti-bonding molecular orbitals is fundamental to the theory of chemical bonding, and is long established by spectroscopic observations. One implication is that changing the energy splitting and the electron occupancy of these orbitals will change the bond strength. Now, an international team of researchers has shown this principle in action using the latest technology for producing bright x-ray beams.
Martin Beye of the DESY Photon Science centre in Hamburg, Germany, and his collaborators, have used pulses from the Linac Coherent Light Source (LCLS) free electron x-ray laser in California, US, to probe changes in the bonding and anti-bonding orbitals formed by the chemisorption of oxygen atoms on the surface of ruthenium metal. The researchers ‘activate’ the Ru–O bond by optically pumping electrons from the metal into the anti-bonding level. Using x-ray absorption (XAS) and emission (XES) spectroscopy, they can follow the bond’s consequent weakening and lengthening. Activation of this bond by thermal energy is crucial to the conversion of carbon monoxide to CO2 in catalytic converters.
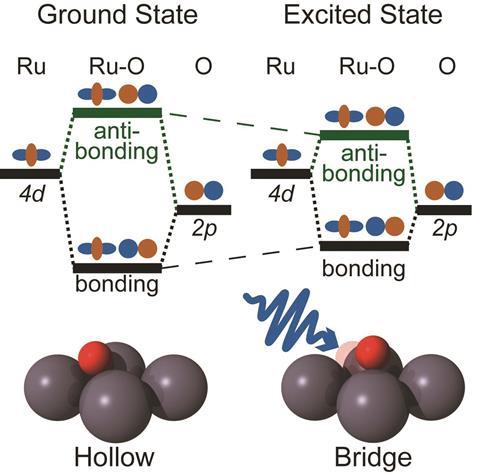
The Ru-O bond results from overlap of the oxygen 2p orbitals with the ruthenium 4d orbitals. In standard molecular-orbital bonding theory, the energy splitting of the orbitals is a measure of the bond strength. In the equilibrium chemisorbed state, only the bonding orbitals are occupied by electrons. But the researchers can populate the anti-bonding states by exciting electrons into it using a very short pulse of an optical laser. The light is absorbed by the metal, boosting its electrons from the valence to the conduction band, from where some can enter the anti-bonding levels of the Ru–O bond.
Beye and colleagues can follow the resulting weakening and lengthening of the Ru-O bond by using the LCLS’s ultrashort x-ray pulses, lasting just a few tens of femtoseconds (10-15s), to pump electrons from the oxygen 1s orbitals into the bonding and anti-bonding orbitals, monitoring the consequent absorption and emission of x-rays. The relative energies of these transitions shift when the anti-bonding levels are populated, showing that the energy splitting has decreased.
It’s only a transient effect: the anti-bonding electrons are quickly shunted back onto the metal, probably in less than a femtosecond for any given electron. The researchers see a return to the original energy splitting just 10 picoseconds (10-11s) after laser excitation of the metal. Weakening of the Ru-O bond makes it longer, and once the electrons exit the anti-bonding states this leaves the bond stretched relative to its equilibrium state – so that it subsequently vibrates.
Beye says that the results could improve the understanding of reaction pathways, and perhaps ultimately lead to the design of better catalysts – for example, where oxygen is more weakly bound in the first place, so that activation isn’t necessary for the oxidation of CO to proceed.
‘This is a benchmark method-development paper, and heralds things to come,’ says chemical physicist Adam Kirrander of the University of Edinburgh in Scotland. ‘It is a strikingly elegant approach, since the XAS and the XES probe the occupied and unoccupied orbitals in one single experiment,’ he says, adding that this sort of information could become useful for intelligent design of molecular complexes with specific target properties.
‘The next step is to extend the method to more complex molecules,’ says Beye. He thinks it might become possible to use an x-ray laser to selectively excite certain orbitals, and thus ‘to stimulate specific chemical reactions and learn about reaction pathways and how to tune them’.










No comments yet