Collective bonding continues to divide opinion
By  Hugh Ryan2024-02-14T10:49:00
Hugh Ryan2024-02-14T10:49:00
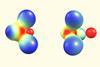
New research supports counterintuitive explanation, but debate is far from settled
When chemists first synthesised NaBH3− back in 2019, initial research suggested a dative bond connected its sodium monoanion with a borane Lewis acid. Theoretical chemists, however, quickly established that the bonding was not that simple.