The origin of the peculiar thermal expansion of a mineral called cordierite has been revealed using lattice dynamics and molecular dynamics simulations.1 Although this ceramic is used in several high-temperature applications like catalytic converters for vehicles, the mechanism behind its tiny thermal expansion has remained unclear. Now, a team working with Martin Dove from Sichuan University in China has unlocked the mystery.
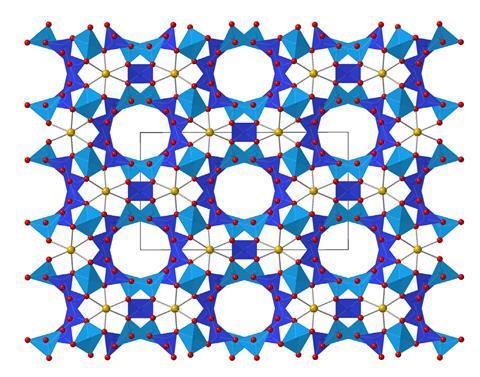
‘The researchers have proposed a theory that allows them to explain why cordierite shows very small overall expansion and negative thermal expansion (NTE) along one direction,’ says Cora Lind-Kovacs at Toledo University in the US, who wasn’t involved in the study. The new results could be used to design materials with interesting thermal properties.
NTE compounds have been studied extensively in the last 30 years, with several materials being discovered and characterised during this time. Although cordierite’s unusual properties have been known for even longer, Dove and colleagues say that recent advances in the field haven’t been used to understand the origin of thermal expansion in this important material. ‘There has been one recent attempt,2 but this did not reproduce the experimental results,’ the researchers say.
Cordierite is an anisotropic material, so its properties vary depending on the direction of measurement. There are three mechanisms used to describe NTE in anisotropic compounds. The first is known as the tension effect and results from atom displacements in the structure, the second is a hinge mechanism, where expansion along one axis automatically causes contraction along a perpendicular axis and the third arises from the existence of a phase transition.
Dove’s team found that the thermal expansion of cordierite could only be explained by a combination of a tension effect and a hinge mechanism. ‘This leads to very small positive expansivities in two directions and negative expansivity in the third,’ they report. To understand the details of the system, the scientists carried out molecular dynamics simulations at elevated pressure and analysed the flexibility of the structure.
‘The lattice dynamical calculations performed by the researchers take into account how atoms in cordierite can vibrate, and how the movement of any given atom will affect all other atoms in the crystalline material,’ explains Lind-Kovacs. ‘They didn’t only consider “phonon modes”, which are inherent vibrations for a given structure, but also the elastic properties of the mineral. With these methods, they could reproduce cordierite’s expansion behaviour well for the first time.’
‘This work shows the very strong link between mechanical aspects and thermal aspects,’ adds Joseph N Grima at the University of Malta. ‘That’s why it’s ideal that you look at these properties in a holistic manner.’
Similar behaviour was observed in layered perovskites in 2017.3 ‘While the joint action of both of these mechanisms has been explored in simpler model systems, such as those based on layered perovskites, the complexity of the cordierite structure, coupled to its technological applicability, make the findings of Dove and Li particularly significant,’ comments Mark Senn from the University of Warwick, UK, who participated in the 2017 study. ‘This could pave the way for computational-driven design of improved compositions where thermal expansion coefficients are tailored for particular high- or low-temperature applications.’
On 29 January 2025, reference 2 was corrected.
References
1 MT Dove and L Li, Matter, 2025, 8, 101943 (DOI: 10.1016/j.matt.2024.101943)
2 Y Li et al, J. Am. Ceram. Soc., 2018, 101, 4708 (DOI: 10.1111/jace.15708)
3 C Ablitt et al, npj Comput Mater., 2017, 3, 44 (DOI: 10.1038/s41524-017-0040-0)









No comments yet