Copper catalysis overcomes double bond trouble
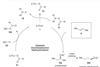
Stubbornly stable unactivated internal alkenes become chiral tertiary amine precursors
Stubbornly stable unactivated internal alkenes become chiral tertiary amine precursors