
This year’s chemistry Nobel prize was awarded to three scientists working in the field of protein design and structure prediction. One half of the prize was awarded to David Baker at the University of Washington in Seattle, US, while the other half was awarded to Demis Hassabis and John Jumper, both from Google DeepMind, based in the UK.
Why is protein structure worthy of a Nobel?
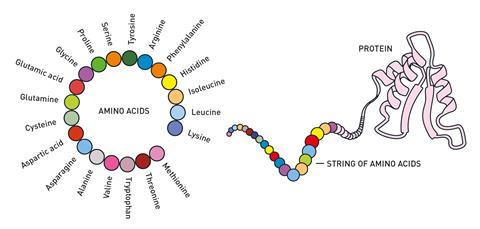
We have known for a long time that proteins are the chemical tools of life – there are many different types of protein that all have different roles in our bodies. Each protein is made of a string of amino acids that folds up into a specific 3D shape, or structure, and each protein’s function is closely related to that shape. Knowing a protein’s structure helps us understand how it works and for decades scientists have been working on ways to figure out protein structures, which has presented many challenges along the way.
In the 1950s, the development of x-ray crystallography enabled researchers to obtain the first 3D structures of proteins. John Kendrew and Max Perutz were awarded the Nobel prize in chemistry in 1962 for that work. Other experimental methods such as NMR and cryo-EM have since been added to the toolkit and researchers have now determined the structures of around 200,000 proteins.
In 1972, American biochemist Christian Anfinsen was awarded the Nobel prize in chemistry for his discovery that it is the sequence of amino acids that determines the way the polypeptide chain folds itself and that no additional genetic information is required. That means it should be possible, in theory, to predict the shape of a protein just by knowing its amino acid sequence.
This finding led to 50-year-long quest to find a way to predict the 3D structure of a protein from its amino acid sequence – but the number of theoretically possible conformations of a protein is, in short, astronomical.
This so-called ‘prediction problem’ became the great challenge of biochemistry and led to the launch of a project, turned competition, in 1994 called Critical Assessment of Protein Structure Prediction (CASP) which aimed to speed up discoveries in the field. However, it was many years before a significant breakthrough was made.
This year’s award recognised two different discoveries – why are they sharing the award?
The work of these three scientists is closely interlinked. Hassabis and Jumper used artificial intelligence (AI) to predict the 3D structure of a protein from its sequence alone. Meanwhile, Baker developed computational methods that could solve the inverse problem: starting from a protein with a particular structure, figuring out what sequence it would have. That enabled him to create entirely new proteins that did not previously exist.
All of this work builds on the decades and decades of research – and chemistry Nobel prizes – on understanding the structure of proteins.
What did the Laureates actually do?
In the 1990s, Baker began to explore how proteins fold. Using these insights he developed Rosetta: computer software for predicting protein structures.
Initially Rosetta was used to convert amino acid sequences into structures, but following the 1998 CASP competition, Baker and his team decided to use the software in reverse; a technique which eventually led them to create completely novel proteins from scratch, also known as de novo design.
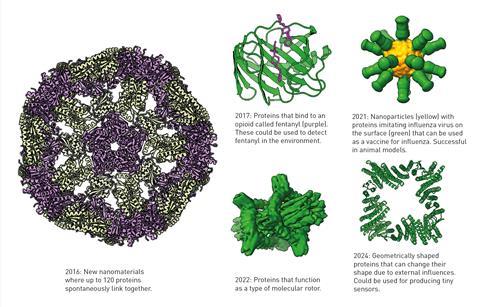
To do this, they drew a protein with an entirely new structure and had Rosetta work out which type of amino acid sequence would result in that protein. They then introduced a gene that coded for their proposed amino acid sequence into bacteria, which produced the novel protein,Top7. Using x-ray crystallography, they were able to determine that the protein they had made had a structure very close to the one they had initially designed.
The work of Baker and his colleagues was published in 2003 and the code for Rosetta was released to the global research community to enable ongoing development of the software and new applications.
In 2010, Hassabis, a British computer science and AI researcher, founded DeepMind Technologies. DeepMind initially developed AI models for popular board games, and following its acquisition by Google in 2014 it achieved a machine-learning milestone when its AlphaGo program defeated the world’s best Go player in 2016. The company went on to construct a computer program based on a convolutional neural network – called AlphaFold.
In 2018, AlphaFold left the rest of the field behind at the 13th CASP competition, reaching 60% accuracy for its predicted protein structures. But getting to higher accuracies presented a new challenge.
Enter Jumper, a researcher with creative ideas about how to improve AlphaFold. Together, Jumper and Hassabis co-led the work that led to AlphaFold2 in 2020, aided by Jumper’s knowledge of proteins and the innovation behind an enormous breakthrough in AI – neural networks called transformers – which could find patterns in huge amount of data more flexibly than ever before.
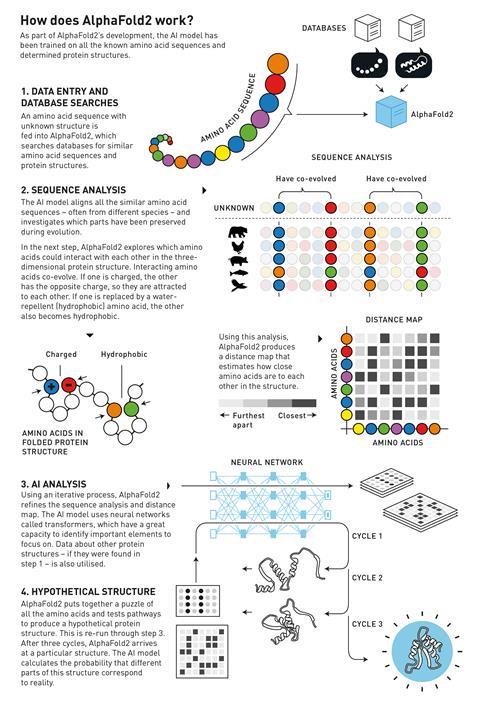
When an amino acid sequence with an unknown structure is fed into the programme, it searches the database for similar amino acid sequences and protein structures. The network then creates an alignment of similar sequences, sometimes from difference species, and looks for correlations between them as well as possible interactions between amino acids. From this information AlphaFold2 can then iteratively refine a distance map – which tells you how close two amino acids are to each other in space – and sequence analysis. Finally, it then converts all that information into a 3D structure.
Now AlphaFold has more than 2 million users and has resulted in the prediction of 200 million protein structures.
What are the applications of this work?
Because of these breakthroughs, most monomeric protein structures can now be predicted with high fidelity, and large databases of hundreds of millions of structures have been created as a result. Proteins are such a key component of our biology that being able to design them and predict their structures opens up potential applications in pharmaceuticals, nanomaterials and rapid development of vaccines, as well as many others.
Does this mean the end of experimental work in this area?
There’s no doubt that the development of AI protein structure prediction tools like AlphaFold represent an important milestone in structural biology, but they are not a replacement for experimental structure determination. Experimentally determined structures are still superior to predictions, and they will also be needed to generate the training datasets for the next generations of AI tools, as well as being used to assess the performance of those tools in predicting structures.
One example of the ongoing need for experimental approaches is in drug design. Although determining a protein’s structure may help generate ideas about what compounds to make next, there are many other factors regarding the biological activity of proteins to consider, such as pharmacokinetics, metabolism and toxicology, that can not currently be solved using AI.
It is much more likely that the future of structural biology will be in integrating high-throughput experimental studies with AI, not replacing it.














No comments yet