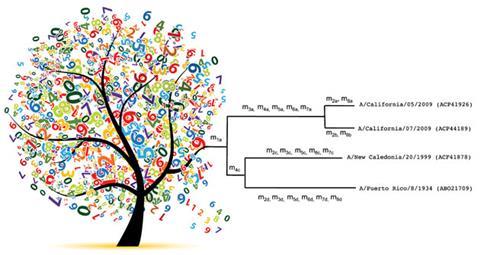
Evolutionary or phylogenetic trees are used to visualise the relationships between several or many different organisms, usually based on differences in their genes. The approach is useful for identifying and classifying microbes, for example, showing how a new strain of pandemic flu virus is related to known strains.
Kevin Downard and colleagues at the Universities of Sydney and New South Wales in Sydney now show that similar results can be obtained by using mass data for protein components – either from lists of theoretical peptide masses or from the digestion and analysis, by mass spectroscopy, of real virus proteins. The authors say their ‘mass trees’ can be built ‘more rapidly and directly’ than those generated from sequence data.
Using theoretical mass data for the haemagglutinin protein – a flu protein the virus employs to bind to human cells – the researchers created a mass tree for 250 flu virus strains from different countries between 1918 and 2012. When they compared the tree with one built using genetic sequence data, it was about 60% similar, as judged by a tree-comparing algorithm. The fewer virus strains they used, the more similar their trees became to those created in the traditional way.
When the tree was built using mass spectroscopy data from the digestion of haemagglutinin proteins, one 2003 strain from Hong Kong and a 2005 strain from Turkey were grouped differently to those in the equivalent phylogenetic tree. However, the researchers say the results are largely similar. ‘Despite these minor differences, the topology of the trees is highly conserved,’ they write.
‘If they can really do this rapidly, you can imagine it could be useful in a flu outbreak, when you’ve got an epidemic spreading,’ says Vincent Moulton, a phylogenetics expert at the University of East Anglia, UK. ‘You might need to do this really fast and then later you might come back and reanalyse [the virus] with sequencing and dig deeper.’
Moulton says any mass-based method is more likely to be complementary than a replacement for traditional genetic sequencing approaches. ‘I certainly think it would be good to have a method that would be independent of sequencing so we could check our results in different ways,’ he says. But he adds that the cost of genetic sequencing is going down all the time, so it might not always be cheaper to build evolutionary trees using mass data.





No comments yet