Hypervalent iodine reagent’s aversion to conversion
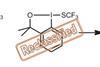
A case of mistaken identity inspires a closer look at the structural stability of Togni reagent I
A case of mistaken identity inspires a closer look at the structural stability of Togni reagent I