





Despite knowing next to nothing about chemistry or biology, a neural network can make a good stab at one of the toughest problems in biochemistry – predicting how a protein folds simply by looking at its amino acid sequence. The machine learning algorithm is a million times faster than other prediction programs, making it a hopeful to win next year’s worldwide protein folding championships.
It is straightforward to analyse a protein’s amino acid sequence, but uncovering its 3D structure is not. Although there are a number of methods – nuclear magnetic resonance, x-ray crystallography and cryo-electron microscopy – they remain laborious and costly. This is part of the reason that there are only around 140,000 structures in the Protein Data Bank – a tiny fraction of the estimated 1012 natural proteins.
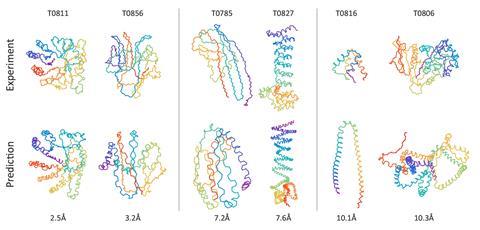
For almost half a century, scientists have been trying to predict how an amino acid sequence twists and folds into a functioning protein. Now, Mohammed AlQuraishi from Harvard University, US, has unleashed a machine learning algorithm on the task. His neural network beat every prediction program that topped one of the last six protein folding world championships prior to 2018 – the biennial Critical Assessment of Techniques for Protein Structure Prediction (Casp).
Tested against challenges set for Casp since 2006, the algorithm is more accurate – by a small margin – than other predictors in the novel folds category. Structures in this group are very different from known ones, making it difficult for software that relies on comparisons with known proteins.
While other programs take hours or even days to perform their simulations of protein folding, AlQuraishi’s algorithm does the same thing in just milliseconds. ‘People search databases of proteins, extract fragments, do various kinds of simulations to minimise physics-based energy functions – very complex, usually millions of lines of code,’ AlQuraishi explains. ‘The idea was to take these very complex pipelines and reformulate them as a single neural network.’
The model knows very little about physics and chemistry, says AlQuraishi, though it respects local geometry and will not put two atoms on top of each other. It learnt about proteins by looking at between 10,000 and 50,000 sequences and their structures for a few months.
AlQuraishi was surprised to find the algorithm had realised that long amino acid chains fold into helices and pleated sheets – the most common structures in proteins. ‘The neural network has learned this on its own, despite not ever being told about the existence of secondary structure,’ he explains.
‘I think the key point about his approach is the fact that it’s fully differentiable,’ says protein folding expert Alberto Perez from the University of Florida, US. This, he explains, makes it easier to design proteins from scratch – simply reverse the process and predict which amino acid sequence produces a desired 3D structure.
Perez thinks AlQuraishi’s neural network could also improve his own simulations. ‘The machine learning is able to get very good overall folds of the protein and then the physics-based approaches are able to refine the details of the structures.’
In 2020, AlQuraishi will enter his neural network into the next protein folding challenge, potentially competing against Google’s artificial intelligence that made waves in last year’s Casp. ‘We’re all expecting great things for next Casp,’ says Perez. ‘[AlQuraishi’s algorithm] could be a major player.’
References
M AlQuraishi, Cell Systems, 2019, 8, 1 (DOI: 10.1016/j.cels.2019.03.006)













No comments yet