Program that automatically interprets NMR spectra is boon for structure elucidation
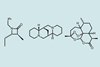
Raw NMR data takes 60 seconds, rather than eight hours, to go from spectrometer to structure
Ever struggled to assign your NMR spectra? Can’t tell which diastereomers you have made? Help is here. Researchers in the UK have developed a program that automatically analyses 1H and 13C NMR data, assigns the peaks, and suggests which structure is most likely from a set of candidates.