Subtle forces yield profound effects on heavy element bonding
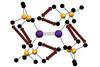
Calculations on dispersion forces shed light on interactions between heavier main group elements
Calculations on dispersion forces shed light on interactions between heavier main group elements