Viewpoint shift designs drug binding proteins from scratch
By  Andy Extance2020-09-07T13:30:00
Andy Extance2020-09-07T13:30:00
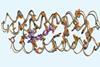
New method focuses on the groups touching amino acid side chains
US chemists have developed an approach that could simplify the currently extremely difficult process of custom-designing proteins for a specific job. Nick Polizzi and Bill DeGrado from the University of California, San Francisco (UCSF) used thousands of protein structures in the Protein Data Bank (PDB) to define a new structural unit. Rather than worrying about the exact positioning of side-chains of each amino acid residue in a protein, they shift focus to the chemical groups each residue is in contact with. Using their method they designed two different proteins from scratch that recognise the blood thinner drug apixaban.