(−)-Gelsemoxonine
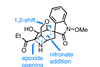
Harnessing the power of the nitro group
Of all the chemical moieties found in natural products, nitro groups are among the least common. Dawei Ma’s group at the Shanghai Institute of Organic Chemistry, China, have completed total syntheses of four members of the ever-popular gelsedine class of alkaloids using a reaction cascade involving an alkyl nitro group that’s essential for both reactions and is somehow then carried through the entire natural product synthesis until its reduction in the penultimate step.