By Chris Nawrat2016-08-08T18:20:00
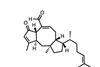
When it comes to cascade reactions, radicals are king of the ring-formers