

Adding fluorine to drug molecules can be tricky, but is often worthwhile. Rachel Brazil talks to the chemists trying to tame the ninth element
-
Significance of fluorine in pharmaceuticals: Fluorine is a crucial element in drug design due to its ability to enhance the properties of drug molecules, such as increasing potency and stability. Approximately 40% of new pharmaceuticals contain fluorine, despite the challenges in creating fluorine–carbon bonds.
-
Innovative fluorination methods: Researchers are developing new methods to incorporate fluorine into drug molecules more efficiently and sustainably. These include photocatalysis, flow reactors, and biocatalysis, which aim to reduce the use of harsh reagents and minimise fluorinated waste.
-
Environmental concerns: The production and use of fluorinated compounds raise environmental issues, particularly with persistent pollutants like PFAS. There is ongoing research to find greener fluorination techniques and to manage the environmental impact of fluorinated drugs.
-
Future directions: The article highlights the potential for new technologies to revolutionise fluorination processes, making them more sustainable and reducing the environmental footprint. Researchers are also exploring ways to selectively add fluorine to drug molecules to balance efficacy and environmental safety.
Summary generated by AI and checked by a human editor
‘Fluorine, it’s a magical element,’ says Rob Young, a drug discovery consultant and former GSK chemist. ‘[Adding fluorine is] the simplest change you can make to the architecture of a drug compound, but can have a very profound effect.’ About 40% of new pharmaceuticals contain fluorine and 25% of all those on the market, including commonly prescribed drugs like atorvastatin (Lipitor) and fluoxetine (Prozac). The drawback is the chemistry involved – creating fluorine–carbon bonds often requires harsh, toxic reagents or expensive catalysts. These reactions can also create fluorinated waste that by some definitions will be classed as polyfluoroalkyl substances (PFAS), the harmful ‘forever chemicals’ that accumulate in the environment and are currently under regulatory scrutiny.
Of course, chemists have been looking for alternative methods that will ease the production of fluorinated drug molecules and agrochemicals. They are also looking to overcome some of the current limitations. ‘The form that fluorine takes in these molecules is inherently limited by the synthetic techniques [available],’ says chemist Julian West from Rice University in Texas, US.
The power of fluorine stems from its small size – not much bigger than hydrogen – and strong electron-withdrawing properties. A fluorine can favourably shift the pKa and lipophilicity of a molecule. ‘Even if you just put it into a phenyl ring … it can have quite a profound effect on potency,’ says Young. The presence of a carbon–fluorine bond is also able to block metabolic ‘soft spots’ in drug molecules, sites where the drugs would otherwise be broken down inside the body. ‘When you have a drug molecule with the fluorine in, you shut down that metabolic pathway and the lifetime of that drug in the body is just longer,’ explains chemist Tim Noel from the University of Amsterdam in the Netherlands, who has designed new fluorination methods.
![Chemical structure of atorvastatin ((3R,5R)-7-[2-(4-Fluorophenyl)-3-phenyl-4-(phenylcarbamoyl)-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoic acid) and prozac, also known as fluoxetine (N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]propan-1-amine)](https://d2cbg94ubxgsnp.cloudfront.net/Pictures/web/y/o/e/drugswithfluorine_65474.svgz)
The electron-withdrawing properties that allow fluorine to improve drug performance also make it difficult to introduce it into drug molecules and difficult to get exactly where you want it within a molecule. There are some highly oxidative commercial reagents to do this. One of the first was Selectfluor – a derivative of the bicyclic tertiary amine base 1,4-diazabicyclo[2.2.2]octane which provides an electrophilic source of fluorine. Others, like diethylaminosulfur trifluoride, provide a route to nucleophilic fluorination. Other methods use expensive metal catalysts. West says there is an over-reliance on adding trifluormethyl groups to drug molecules using reagents like trifluoromethyl iodide. ‘We don’t have a corresponding library of different kinds of other organofluorine fragments … we don’t have the reagents that can actually install those fragments.’
Photocatalysis and going with the flow
West has developed a method to hydrofluorinate alkenes in milder redox-neutral conditions than was previously possible, starting with fluoroalkyl carboxylic acids. The reaction uses only a cheap iron salt such as iron (iii) diacetate as the photocatalyst and an organic thiol hydrogen donor. On photo-excitation the iron is reduced and a carboxyl radical forms, which leads to the loss of carbon dioxide, leaving a fluoroalkyl radical to react with the alkene. ‘[The iron photocatalyst] is able to decarboxylate things that are incredibly hard to decarboxylate … you just pull out an electron, [which is difficult] if you have these three fluorine atoms on the other side of this carboxylic acid,’ explains West. The reaction is completed with the thiol re-oxidising the iron and forming a thiyl radical. This then donates a hydrogen atom to the carbon radical formed on adding that perfluoroalkyl group to the alkene, leaving a new fluorinated molecule.

The reaction is attractive because it can be carried out at a late stage in the synthesis of a bioactive molecule if an alkene group is present (which it commonly is in drug molecule intermediates), which means less fluorine waste created in previous stages of any multi-stage synthesis. Plus West says the reaction works for all of the perfluoroalkyl groups that they tried, including mono, di and tri-substituted. ‘You could generate a library of organo-fluorine drug candidates just using this single reaction … basically any carboxylic acid that you could buy with fluorines is open.’
The one drawback is the fluoride always adds to the less substituted carbon, due to the stability of the free-radical produced in the alkene addition. The alternative regiochemistry is ‘absolutely inaccessible’ using this method. ‘That’s one potential challenge,’ concedes West.
Another method is being developed by Noel and collaborators, including chemists at AstraZeneca. He has found that in flow reactors he can add trifluoromethyl groups attached to a sulfur, nitrogen or oxygen atom using only a caesium fluoride salt as the source of fluorine. The trifluoromethyl–heteroatom motif is commonly used in drug molecules.
Flow reactions, which take place in a continuous stream inside a closed system of tubes, improve the contact between reactants as well as keeping toxic intermediate species safely contained. Starting with an imidoyldichloride (Cl2=NR) molecule (or thiophosgene (CSCl2) or diphosgene (ClCO2CCl3)), Noel’s reaction is based on two successive chlorine–fluorine exchanges and the addition of a final fluorine to form an anion which is immediately reacted with an electrophile to form a new carbon–heteroatom bond next to the trifluoromethyl group.
The reaction only takes 10 minutes. ‘Because we are working in a packed bed reactor where there is a rich source of fluoride, it immediately pushes the equilibrium towards the anion so the risks that are associated with handling [toxic] intermediates are also immediately tackled,’ says Noel. The advantage over existing methods is the ‘on-demand’ fluorination which cuts out the need for perfluoroalkyl precursor reagents. ‘We really have only the CF3 group in the latest stage that is possible,’ he adds, which limits further generation of fluorinated waste.
‘We cannot replace every single [fluorination] reagent yet, but I think it’s a good start.’ says Noel. His group are now working on extending the method using other anions and are also looking at applying the method to high throughput experiments. ‘Our hope is that this will become a go-to technology.’
Biocatalysis
Another obvious direction to avoid harsh reaction conditions and perfluorinated starting reagents is biocatalysis. But there are few enzymes designed to form carbon–fluorine bonds in nature. One of the rare examples known as fluorinase or 5’-fluoro-5’-deoxyadenosine synthetase (FDAS) was isolated from a class of soil bacteria and characterised by University of St Andrews chemist David O’Hagan in 2002. Since then, O’Hagan has been looking at how we might exploit this enzyme to produce organofluorine compounds. ‘Progress has actually been quite slow, truth be told,’ admits O’Hagan. ‘It’s been quite a challenging area.’
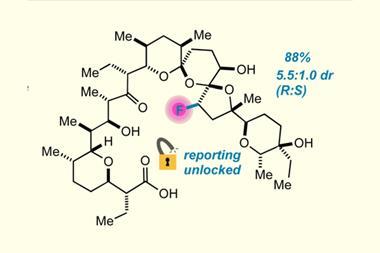
There are a handful of fluorinated compounds in nature – fluoroacetate being the most common. ‘It’s toxic to grazing animals so the plant seems to produce it to fend off animals grazing on them,’ explains O’Hagan. It is generated from the nucleophilic substitution of S -adenosyl-l-methionine to form 5-fluorodeoxy adenosine (FDA) catalysed by fluorinase, and FDA is then biotransformed to other fluorometabolites over multiple steps. In 2010 O’Hagan added the gene for fluorinase to the marine bacteria Salinispora tropica, replacing the similar chlorinase, and was able to induce the production of fluorosalinosporamide – a fluorinated version of a potential anticancer natural product.
![Structure of fluorinated natural product fluorosalinosporamide ((1R,4R,5S)-1-((S)-((S)-cyclohex-2-en-1-yl)(hydroxy)methyl)-4-(2-fluoroethyl)-5-methyl-6-oxa-2-azabicyclo[3.2.0]heptane-3,7-dione)](https://d2cbg94ubxgsnp.cloudfront.net/Pictures/web/y/o/e/naturalproductlikething_38740.svgz)
But O’Hagan says there is still a ‘a way to go’ in usefully using the enzyme. One big problem is that the fluoride ion can be toxic to cells at high concentrations and so cells have mechanisms for removing it. ‘What has emerged for success at this is you have to somehow or other control fluoride ion concentration within the cells,’ says O’Hagan, which means blocking the channels through which fluorine is exported.
The other major issue is the kinetics of the reaction. ‘It’s quite a slow process,’ says O’Hagan. Some labs are using directed evolution to try and improve the kinetics but so far nothing significantly faster has been found. Part of the problem is desolvating the fluoride ion. ‘The fluoride ion in water is highly hydrated, and any enzyme has to dehydrate the fluoride and the energetics of that process are high,’ O’Hagan explains.
O’Hagan has also been studying nucleocidin, another fluorine-containing molecule produced by the Streptomyces calvus bacteria with antibiotic properties. It’s based on the nucleoside adenosine substituted with a fluorine atom and a sulfamyl ester, and is not synthesised from fluorinase but via an as-yet undiscovered enzyme. ‘The community has been looking at fluoro-nucleosides for all sorts of diseases, including HIV [and] hepatitis C,’ says O’Hagan. The recent hepatitis C antiviral blockbuster drug Sovaldi (sofosbuvir) has a fluorine at the ribose 2′ position. But nucelocidin is uniquely fluorinated at the 4′ position. ‘It’s the most challenging position [to substitute],’ explains O’Hagan. ‘Having the biotechnology to do that would be aspirational.’
Other groups are finding ways to incorporate non-natural fluorinated motifs into molecules using different enzymatic pathways. A team from the University of Illinois at Urbana–Champaign in the US, led by chemist Huimin Zhao, recently used two enzymes, the first to photoinduce a carbon-centered fluoroalkene radical from trifluoroiodoethane, and the second, an acyltransferase to react the radical with an alkene, in this case a vinyl arene. As with all enzymatic reactions the mechanism is enantioselective, which can be a big advantage. But as O’Hagan points out, ‘if you get an enzyme for one enantiomer, it’s not obvious how you get the other enantiomer – [that becomes] a new project!’
Going back to the source
Perhaps the biggest breakthrough in fluorination came in 2023, with a method that goes back to the source of all fluorine-containing reagents – the mineral fluorspar, calcium fluoride. At the University of Oxford in the UK, Véronique Gouverneur decided to find a way to make it more reactive. It’s insoluble in water and organic solvents and therefore needs to be treated with concentrated sulfuric acid at high temperature to produce toxic hydrogen fluoride. All fluorochemicals, including pharmaceuticals and agrochemicals, start from this dangerous reaction.
Gouverneur’s method, developed with colleagues at University College London and Colorado State University in the UK and US, takes its inspiration from the biomineralisation that occurs in forming teeth and bone. She combined fluorspar with powdered potassium hydrogen phosphate (K2HPO4) in a ball mill and a mechanochemical process produced two new crystalline phases, K3(HPO4)F and K2−xCay(PO3F)a(PO4)b. The powdered mixture, which Gouverneur calls Fluoromix, can create bonds between fluorine and either carbon or sulfur. Gouverneur’s group has used it to make 50 different fluorochemicals by directly substituting other halogenated bonds. They found the method does have some limitations, and wouldn’t work for fluoroarenes, which commonly feature in drug molecules.
In her latest work Gouverneur ‘demonstrated that novel and many commonly used nucleophilic fluorinating reagents can be prepared directly from CaF2’. The new chemical method treats fluorspar with electron-accepting boric acid (B(OH)3) in the presence of oxalic acid which is capable of sequestering the Ca2+ ion. The low temperature aqueous reaction starts with the formation of tetrafluoroboric acid HBF4 which acts as a fluorinating agent, exchanging chloro or amine groups to synthesise fluoroarenes and a variety of other fluoro-substituted molecules. Gouverneur and her team have achieved similar results replacing the boric acid with silicon dioxide, which has similar electron accepting properties.

Fluoromix is now being commercialised by Oxford spin-out company FluoRok and could lead to a huge paradigm shift in how all fluorochemicals are manufactured. If you can go to the source material, that’s a huge benefit, says Noel. ‘The fewer intermediates you have, the fewer problems you will have generating wastes, so it’s very exciting.’
Gouverneur hopes this is the start of a move towards a more circular and sustainable fluorochemicals industry, where it’s not only simple to make carbon–fluorine bonds but easier to break them too. ‘The task ahead of us to make this industry “easier and more sustainable” is substantial, and an approach looking at all these processes individually is time-consuming and demanding,’ she says. Part of the solution will be to develop ways to use waste fluorochemicals as a fluorine source because, as Gouverneur warns, fluorspar deposits on earth are finite and depleting rapidly.
Environmental impacts
This ambition is particularly important given the environmental impact of fluorinated compounds – most notably perfluorooctanoic acid (PFOA), and perfluorooctane sulfonic acid (PFOS), which have both been associated with toxicity in animals and humans at sufficient levels, leading to liver damage, thyroid disease, obesity, fertility issues and cancer. Another acutely toxic fluorinated molecule is trifluoroacetic acid (TFA), which is highly persistent in water courses with no degradation pathways. A recent study showed that drugs containing trifluoromethyl groups like fluoxetine (C17H18F3NO) release TFA when broken down. ‘CF3 groups are really popular [in drug molecules] because it’s like putting a bit of Teflon in your molecule,’ says Young. Even though they only have one polyfluorinated carbon, by some definitions they are PFAS – albeit the shortest possible.
Current European proposals to ban the use of PFAS by 2027 could impact medicine manufacture, according to the European Federation of Pharmaceutical Industry Associations. They say the widest definition of PFAS encompasses 10,000 chemicals, including some of the building blocks for medicines. Another study found the broadest definition would include 360 medicines (such as Prozac and Lipitor).

‘The major source of CF3 is not pharmaceuticals or agrochemicals,’ O’Hagan points out. ‘The major source is coming from refrigerants.’ The most common of these is 2,3,3,3-tetrafluoropropene, also known as 1234yf (CH2=CFCF3). ’They are designed to break down because we don’t want them to get into the atmosphere, but they break down eventually to TFA.’ Young and O’Hagan question whether the much smaller amounts of fluorinated drug molecules produced really pose a risk in comparison to the 34,000 tonnes of 1234yf produced each year.
‘I think there’s a broad concern at the moment that the legislation should be much more nuanced in how it considers CF3’ says O’Hagan. He worries that the current situation leaves pharma and chemical companies developing fluorinated molecules in limbo. Plus, Noel adds that the alternative, without fluorine, means higher drug doses, potentially increasing patient side effects.
West wonders if we have gone too far in fluorinating drug molecules. ‘Can we make these things too stable? Do we have to worry about excretion and long term environmental effects as they accumulate?’ Young agrees: ‘Some drugs overdo it.’ He points to the HIV drug lenacapavir which includes 10 fluorine atoms and only needs to be taken every six months. ‘Do you fancy being given something that’s bioactive and hangs around for that long?’ he asks. Young says this is a debate that ‘needs to be had’. ‘We don’t want to throw the baby out of the bathwater, but what is used should be used judiciously.’
West may have a solution to this – he is looking for a way to add fluorine only in positions on a drug that are known to be vulnerable to metabolic break-down. You could imagine a complex molecule with an eight-carbon chain, he explains, where the molecule is broken down in the body at the fourth carbon. ‘We want to block that, but we don’t want to make some sort of weird PFAS molecule. We just want fluorine at that fourth carbon.’ He says his group has a photocatalytic approach that allows them to do this, which they have not yet published but hope might provide a way to reduce levels of fluorine without sacrificing efficacy.
Ultimately, it will be chemists who provide less toxic routes to fluorination reactions as well as dealing with persistent fluorinated chemicals in the environment. ‘You see people now developing new methods to defluorinate molecules,’ says Noel. ‘I think that the problem will be solved in a couple of years.’ He is sure that moving away from fluorine in drugs is not the solution. The F in pharma is almost certainly here to stay.
Rachel Brazil is a science writer based in London, UK












No comments yet