

Andy Extance charts how research into revolutionary targeted protein degradation therapies is moving from serendipity to strategic discovery
-
Revolutionary drug development: Targeted protein degradation therapies, which involve tagging disease-related proteins for destruction by the proteasome, are emerging as a revolutionary approach in drug development. This method can target proteins that traditional small-molecule inhibitors cannot, offering new treatment possibilities for various diseases.
-
Clinical progress: As of 2024, numerous TPD drugs are in clinical trials, with companies like Arvinas leading the way. Their breast cancer drug, vepdegestrant, has completed a phase 3 clinical trial, showcasing the potential of TPD in treating cancers and other diseases.
-
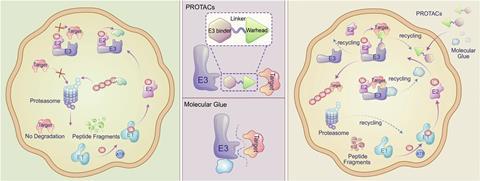
Mechanisms and strategies: TPD relies on two main strategies: heterobifunctional degraders, which link a target protein to an E3 ligase, and molecular glues, which stabilise the interaction between the target protein and the ligase. Both strategies aim to tag the target protein for degradation by the proteasome.
-
Challenges and innovations: Despite its promise, TPD faces challenges such as optimising drug-like properties and identifying suitable target–ligase pairs. Innovations like high-throughput screening platforms and new linker designs are helping to overcome these hurdles, paving the way for more effective and selective TPD therapies.
This summary was generated by AI and checked by a human editor
One of today’s hottest pharmaceutical research areas owes much to alphabetically ordered posters at the 1999 annual Burroughs Wellcome Fund (BWF) scholars’ retreat. Though they both had received a BWF Junior Faculty Award, Craig Crews and Ray Deshaies hadn’t discussed their work before. Positioned together because their surnames followed each other, the pair started an exchange that continued over beer. Crews was studying approaches to bring proteins together. Deshaies was studying how E3 ubiquitin ligase enzymes can label proteins to be dispatched for destruction by the cellular shredder – the proteasome. ‘I could actually drag in proteins of interest and hijack your E3 ligases,’ Crews recalls telling Deshaies.
With colleagues, they demonstrated this targeted protein degradation (TPD) concept in 2001, exploring its potential for several years. In 2008 Crews, at Yale University in New Haven, US, decided to convert this academic concept into a radical new drug modality. Traditional small-molecule inhibitor drugs typically jam a protein’s function like a wrench in the gears. Chopping proteins involved in disease into confetti instead could enable treatments previous approaches could never achieve. Scientists have discovered more than 4000 disease-associated proteins, of which current therapies target around 400. The remainder often have difficult structures to target – but TPD drugs might be able to.
In 2024, many drugs using this approach are now in clinical testing, says Alessio Ciulli from the University of Dundee in the UK: at least 36. Meanwhile immunomodulatory medicine (IMiD) cancer drugs also work by causing E3 ligases to tag proteins for degradation, researchers have been revealing since in 2010. Regulators approved the best known IMiD, thalidomide, to treat the bone marrow cancer multiple myeloma in 2006 without fully understanding how it worked. Ciulli stresses these molecules are a new class, differentiated by their degradation mechanism. ‘It’s really revolutionising how we think about developing a drug,’ he tells Chemistry World.
Among many TPD companies is Arvinas, founded by Crews in New Haven, which has the most advanced new drug candidate. A potential breast cancer drug, vepdegestrant completed a phase 3 clinical trial in November 2024. Arvinas is due to report results in early 2025. Yet, while what Crews and Deshaies outlined at first appears an enticingly simple picture, as researchers look more closely into TPD, its complexity only grows.
Target, tag, destroy
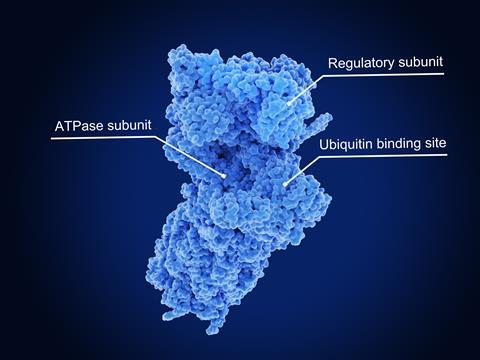
TPD relies on the tens of thousands of proteasomes already in each of our cells, explains Si-Han Chen from Bristol Myers Squibb in San Diego, US. Each proteasome is a cylindrical protein complex with a cap unit on one end that recognises ubiquitin proteins, which mark other proteins for degradation. An unfoldase enzyme in the cap pulls proteins into the cylinder, unfolding them. Inside the cylinder, there are three types of protease enzyme. Each protease grabs a different type of amino acid building block in the entering protein, before snipping the link to the next amino acid. The resulting confetti-like chains are 2–24 amino acids long, Chen stresses, which other enzymes break down before making new proteins with them.
This natural degradation process is crucial for cellular rejuvenation, maintaining protein quality by removing misfolded, damaged proteins from cells, explains Ciulli. The first degraders from Crews and Deshaies used short amino acid chains, known as peptides, to bind with ubiquitin-adding ligases. But peptides don’t enter our cells easily: our bodies break apart their amino acids before they get anywhere near a proteasome. So to make drugs, Crews needed different structures to bind ligases. He worked with Ciulli and others to make more conventionally drug-like molecules, leading to three breakthrough papers in 2012 targeting the von Hippel-Lindau (VHL) ligase. ‘This gave confidence early on that we could get to a more drug-like molecule,’ says Ciulli.
There are two key strategies for hijacking the proteasome to degrade target proteins, explains Paige Mahaney, chief scientific officer at C4 Therapeutics in Massachusetts, US. In one strategy, heterobivalent degraders like those Crews, Ciulli and colleagues developed have a ligand that binds the protein targeted for degradation at one end, and a ligand for an E3 ligase at the other. A linker structure connects the two ligands, Mahaney explains. In the other strategy, molecular glues like the IMiDs bind where target proteins and ligases meet, stabilising or inducing interactions between them.
‘The potential for TPDs is vast and I am excited to see the advancement of the next-gen degraders,’ says Mahaney. Challenges, however, include optimising drug-like properties and identifying suitable target–ligase pairs, especially for molecular glues with shallow binding interfaces.
The recognition in 2010 that IMiDs hijack an E3 ligase ‘was a wonderful validation for’ TPD, says Crews. Such molecular glues are usually small drug-like molecules, yet their discovery has been serendipitous to date, says Crews, which limits rational drug development. By contrast heterobifunctional degraders allow researchers to choose the ligands at each end, offering a more rational, targeted and systematic approach, he adds.
Thalidomide looms
Allan Jordan, vice president of oncology drug discovery at Sygnature Discovery in Nottingham, UK, notes thalidomide’s massive importance in the field. However, it means finding new patentable molecular glues has been challenging, because thalidomide and related structures are so well studied. Researchers therefore approach new molecular glues through ‘unbiased screening’. This involves looking for ligases and proteins that transiently bind, sending the protein for degradation, without intentionally targeting either partner. Scientists then screen small drug-like molecules, examining whether they further stabilise the pair. Sygnature offers a platform to help companies do such screening. ‘We can then see if any of the degraded proteins are therapeutically viable, and we can actually do something with them,’ Jordan explains.

Roland Hjerpe, senior principal scientist in induced proximity therapeutics at Sygnature, says thalidomide’s small size and attractive physicochemical properties also make it an ideal ligase-binding ligand for heterobifunctional degraders. As a result, most heterobifunctionals in clinical testing use thalidomide or related structures, recruiting the ligase it targets, cereblon. Ligands for other ubiquitin ligases, such as VHL, are larger and less favourable for use in drugs. Thalidomide is ‘a very important small molecule’, driving innovations now showing promising results in clinical pipelines, Hjerpe says.
Jordan argues thalidomide helped shift the focus from peptide-based ligands in heterobifunctional degraders to small molecule ligands. That’s largely because cereblon is so promiscuous it ubiquitinates nearly any protein. He recalls a colleague saying at a conference that if degraders hadn’t started targeting such a versatile ligase then this approach might not exist at all.
As a result, in heterobifunctional design, medicinal chemists focus on linkers, which control how ligase and target proteins align, adds Hjerpe. That firstly influences whether the ligases can add ubiquitin tags, he explains. Secondly, linkers also affect cooperativity, where protein–protein interactions aid how the ligase and target protein come together. And thirdly, they can enable ‘chameleonicity’ , where molecules fold up to hide polar groups with unbalanced distributions of electrons inside fatty structures where electrons are more evenly distributed. That can improve cell penetration and bioavailability, but the behaviour is unpredictable. Finally, medicinal chemists must consider how patients’ bodies break down the linker, Hjerpe says.
Optimising linkers helps create a stable interface between the target protein and the E3 ligase, says Crews. That should ensure drugs bind more specifically with the target protein. And whether developing molecular glues or heterobifunctionals, proteomics is an essential tool in TPD, he adds. For example, analysing the entire proteome of cells treated with drug candidates can assess exactly which proteins are degraded, checking for unwanted potential off-target effects.
We’ve been able to degrade almost all of the oncogenic drivers
Ciulli’s team has been involved in rational TPD drug development that focuses on stabilising E3 ligase–target protein interactions. The Dundee researchers captured the first crystal structure of a heterobifunctional degrader molecule bound simultaneously to both its target and E3 ligase. ‘We found that stabilising this complex increases the efficiency of ubiquitination, leading to faster and more potent degradation,’ Ciulli says. ‘If we can actually see the structure of the complex, we can design modifications in a very targeted fashion to really try to favour recruitment.’
Building on such knowledge, Ciulli’s lab worked with German drug company Boehringer Ingelheim to develop prototype degraders for the protein K-Ras containing ligands for VHL. K-Ras is the most commonly mutated protein in human cancers, where it tells cancer cells to grow. Existing approved drugs target just one specific amino acid mutation, but TPD can fight many cancer types with other K-Ras mutations. ‘We’ve been able to degrade almost all of the oncogenic drivers,’ Ciulli says, offering exciting new possibilities for treating cancer. Although VHL ligase ligands are less drug-like, he adds, at least four drug candidates use them.
Rewriting rules
Another key advantage of heterobivalent degraders is that they ‘liberated’ medicinal chemists, says Mahaney. Researchers felt constrained by guidelines widely used to define the structures of drugs people take in pill form, known as Lipinski’s rule of five, named after their proponent, Pfizer’s Chris Lipinski. Linking ligands for two different proteins usually gives larger molecules than allowed by the rules. As Lipinski warned, such molecules often don’t dissolve or cross gut cell membranes as well as smaller ones. Working in heterobivalent degraders has allowed chemists some freedom to move beyond the rule of five, Mahaney says. But they have also found many fundamental principles still apply, such as limiting the number of hydrogen bonds potential drugs might form.
Drug developers can also use TPD to target non-enzymatic proteins, such as transcription factors, regulatory proteins and scaffolding proteins, which were previously considered ‘undruggable’ Crews says. As the field matures, the focus is shifting to discovering ligands for such targets, he explains. Researchers are asking if they can target ‘a nook or cranny on the surface of these undruggable proteins’, says Crews.
Differing TPD design principles are already evolving the field, argues Hjerpe. TPD drugs in clinical trials link previously known protein-binding ligands to E3 ligase ligands. As investment shifts towards exploring less established targets, including those previously untargetable with inhibitors, scientists are looking for novel structures to bind them. One example is central nervous system (CNS) disorders like Alzheimer’s disease, which involves misfolded proteins like amyloid beta and tau proteins aggregating into plaques and tangles respectively. Such aggregating proteins ‘are difficult to remove by using inhibitors’, Hjerpe says.
Sygnature has therefore developed a platform called Charmed to accelerate heterobifunctional degrader discovery. Charmed uses high-throughput chemistry to rapidly generate and biologically screen heterobifunctional molecules, determining if a target is degradable and identifying lead molecules efficiently. Jordan explains that they have structures known to bind to cereblon or VHL pre-attached to linkers pre-dispensed into plastic 96-well plates. Customers bring a ‘warhead’, a chemical structure known to bind to target proteins. ‘Assuming the chemistry is amenable, we can link those together pretty quickly,’ Jordan says. ‘It takes about a week to generate a few hundred degraders. We talk about democratising degraders – a way of making things accessible to smaller companies.’
Charmed’s focus on just two ligases reflects one key challenge for TPD. ‘Compounds that bind to either a ligase or to a target protein are challenging to develop,’ comments Ingrid Wertz, co-founder and chief executive of Lyterian Therapeutics in San Francisco, US. A key example is that despite significant investment in developing good VHL ligands, Arvinas opted for drugs incorporating cereblon-recruiting thalidomide analogues as lead candidates for clinical trials. ‘The TPD community was surprised,’ Wertz says.
TPD must also overcome new challenges posed by unforeseen complexity. For example, when working at Genentech in San Francisco, US, Wertz and Chen were surprised to find the approach was not working as expected. They tested molecules intended to degrade bromodomain and extra-terminal (BET) proteins. In some tests cells were unexpectedly surviving, despite BET being absent. ‘I was really confused,’ Chen recalls.
Beyond cancer
Wertz, Chen and colleagues discovered that peptides formed when the proteasome chopped up BET were killing cells by binding with proteins known as inhibitors of apoptosis. Resistant cells produced fewer such peptides, despite degrading BET. The unexpected mechanism is ‘an additional layer of complexity that we have to pay attention to,’ says Chen. It stresses the need to be sure a TPD drug is working through ‘a suitable modality’, she adds. The presence of cell-killing peptides is appropriate when treating cancer, she explains but not in treating other ailments such as inflammation or neurodegeneration.
The complexity also offers an exciting opportunity, adds Wertz. ‘It allows you to design your drugs intentionally to either generate these peptides if the goal is to promote target cell death, or to avoid generating them if the intent is to promote target cell survival,’ she says.
Protein degradation often leading to different biological outcomes than inhibition is a nuance requiring deeper understanding of target biology and degradation, Mahaney adds. Success depends on optimising entire drug molecules, as subtle changes significantly affect degradation. ‘I think early researchers in the TPD field thought that degradation was straightforward both chemically and biologically,’ Mahaney comments.
It’s also harder to use cell-based and other tests to predict their pharmacokinetics – how drugs move in the body – in animals and humans. ‘Consequently, we must do more experiments in animals,’ Mahaney says. Experienced scientists at C4 Therapeutics provide the knowledge to tackle these challenges, Mahaney continues. Now, their pre-clinical results translate well to performance in clinical trials. ‘What is surprising is how well-behaved these molecules are once they are optimised,’ she says.
These results offer potential for new treatment approaches for CNS disorders and brain tumours
TPD isn’t the only drug strategy seeking to break down target proteins. Small molecule selective oestrogen receptor degraders (Serds) change the shape of receptors that signal cancer cells to grow. In this way, clinically approved Serds elacestrant and fulvestrant recruit an E3 ligase called arkadia or RNF111 that sends the receptors to the proteasome. The current leading TPD drug, Arvinas’ vepdegestrant, is also seeking to degrade oestrogen receptors.
Vepdegestrant is in trials as a first- and second-line treatment for hormone receptor-positive metastatic breast cancer. In July 2022, Pfizer paid Arvinas $650 million (£530 million) up front, plus a $350 million equity investment, to jointly develop it. Arvinas says vepdegestrant is much safer than imlunestrant, a Serd developed by Eli Lilly, also soon to report phase 3 trial results. ‘We have a clean drug with a very strongly trending clinical benefit,’ Arvinas’ chief medical officer Noah Berkowitz told the Guggenheim Securities Healthcare Innovation Conference in November 2024.
Mahaney notes that trials expanding beyond cancer into areas like immunology and neurology are proving TPD works, showing promising efficacy and robust pharmacokinetics. C4 Therapeutics has developed highly selective degraders whose CNS-penetrating ability she says is exciting. Some show significantly better brain exposure than corresponding small molecule inhibitors. ‘These results offer potential for new treatment approaches for CNS disorders and brain tumours, especially given the excellent selectivity that can be achieved,’ Mahaney says.
Three approved molecular glue IMiD therapies show TPD enables successful treatments, Mahaney adds. ‘There has been incredible progress across TPD in recent years based on deeper understanding of how to design small molecular weight molecules that can overcome limitations of existing therapies,’ she says. ‘Now, several degraders are moving quickly through clinical development toward regulatory review. It’s a very exciting time for the field.’
Andy Extance is a science writer based in Exeter, UK












No comments yet