
Clare Sansom uncovers the complex world of the spliceosome, a molecular machine in all our cells
20 years ago, as the Human Genome Project was coming to an end, Ewan Birney of the European Bioinformatics Institute near Cambridge, UK, set up an online sweepstake over one of the burning questions of the day: how many genes are there in the human genome? More than 200 bets were placed, with an average prediction of over 60,000 genes. The final answer came as a surprise, and, perhaps, a disappointment: the winning bet of 25,947, placed by Lee Rowen of the Institute for Systems Biology in Seattle, US, was the lowest of all and still an over-estimate. The number of protein-coding genes in the human genome is now known to be about 20,400: barely more than that of the millimetre-long nematode worm that is beloved of geneticists worldwide as a simple model organism. Other vertebrates have similar-sized genomes.
The puzzle of how a relatively small number of genes can give rise to the vast complexity and variety of vertebrate biology is a complicated one, but its main solution can be found in the path between the genome and the proteome. Almost everyone who has studied high school biology will be familiar with the central dogma of molecular biology: DNA makes RNA makes protein. And this essentially sums up what happens in bacteria: the ‘molecular machines’ of protein synthesis, the ribosomes, grasp hold of the messenger RNA (mRNA) immediately it is synthesised and start making proteins. An electron micrograph of this process will show the mRNA decorated by its ribosomes like beads on a string.
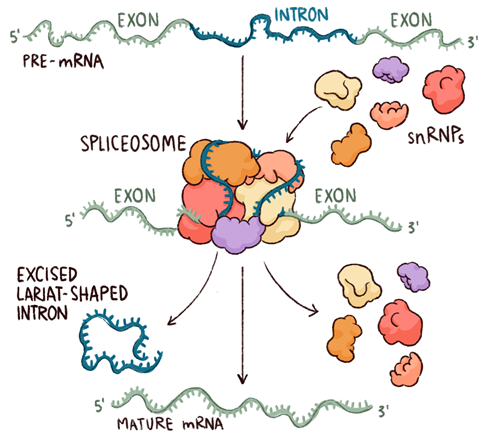
In eukaryotes – that is, all organisms with cells containing nuclei, from amoebae and worms to humans – the same central dogma masks a much more complex process. Messenger RNA is synthesised directly from the DNA making up the genes, as in bacteria, but this is an immature ‘pre-mRNA’ that must undergo successive chemical modifications – and be exported from the nucleus where it is formed – before it can be used in protein synthesis. Two of these modifications, the addition of a single-nucleotide cap to the end of the pre-mRNA where transcription starts (the 5′ end) and a poly-adenosine sequence to its 3′ ‘tail’, are straightforward and universal. It is the third and most complex of these modifications, splicing, that gives rise to the extraordinary expansion of the protein repertoire in vertebrates and, arguably, to their complex biology.
Splicing is the process through which non-coding segments of DNA, known as introns, are removed from pre-mRNA and the remaining exons joined to form one long protein-coding sequence. Almost all eukaryotes use it, but more complex organisms have gene structures that use it more frequently. Alternative splicing, in which different sets of exons from the same gene can be joined to form different proteins, occurs in about 95% of human genes. It has been suggested that the mere 20,000 or so genes in our genomes might produce as many as half a million different proteins.
A structural problem
If you want to understand the mechanism of a biological process, it helps to know the structure of the proteins involved. This has been understood ever since the very first protein structure – of haemoglobin – was published by Max Perutz and his colleagues at the MRC Laboratory of Molecular Biology (LMB) in Cambridge back in 1958. Until recently, however, it seemed impossible that such a complex process as splicing and the molecular machine that drives it, the spliceosome, could yield its secrets to structural biologists.
And one structural biologist will always be associated with the complex and elegant mechanism of this molecular machine: Kiyoshi Nagai, who led the spliceosome group at the LMB. He left his native Japan to work with Perutz as a student visitor in the late 1970s, initially on haemoglobin, and impressed his boss enough to be invited back as a postdoc in 1981. He was given tenure six years later and stayed there for the rest of his life: he was still working as a group leader at his death in September 2019 at the relatively young age of 70.

The LMB is not only known as the UK’s ‘Nobel Prize factory’ – twelve prizes have been awarded to scientists working there, all but three for chemistry – but it is famous as a pleasant and productive working environment. Nagai’s close collaborator Chris Oubridge is another LMB scientist who stayed. He arrived in 1988 straight after his first degree and has been there ever since. ‘I joined the group soon after the lab director, Aaron Klug, had persuaded Nagai to leave the haemoglobin project and branch out into a new research field,’ remembers Oubridge. ‘It was Klug – whose chemistry Nobel in 1982 had been awarded partly for work on DNA–protein binding – who first suggested to Nagai that he work on RNA-binding proteins.’ Whereas DNA almost always forms the famous double-helix structure, RNA structures are much more varied, and very often more fragile. ‘These RNA–protein complexes presented more of a challenge to 1980s structural biologists than DNA–protein complexes, but Kiyoshi rapidly proved that he was up to that challenge,’ Oubridge says.
Nagai’s early work led to the discovery of a novel protein structure associated with RNA binding. This consists of a small beta sheet flanked by two helices on one side and RNA bound to the other. As luck would have it, some of the earliest proteins found to contain this motif – now known as the RNA recognition motif, or RRM – were protein components of the spliceosome, or rather, protein components of parts of the spliceosome. Studying RNA–protein binding in these small structures sparked the interest that carried Nagai’s work forward for the rest of his career.
The structure is energetically unfavourable,so it’s not surprising that it took us so long to obtain enough good crystals and solve the structure
The spliceosome itself is, as molecules go, both huge and hugely complicated. To start with, rather like the ribosome, it consists of subunits that exist independently when inactive and only come together around the nucleic acid to form the active machine. However, whereas the ribosome has two subunits, each comprising many proteins and RNAs, the intact spliceosome includes five so-called ‘small nuclear ribonucleoproteins’ (snRNPs, often pronounced ‘snurps’) and numerous associated proteins. The snRNPs contain RNA that is rich in uracil, the RNA base that replaces DNA’s thymine, and they are named – perhaps unimaginatively – U1, U2, U4, U5 and U6. Nagai began by working out the structures of parts of the U1 and U4 snRNPs, using NMR as well as x-ray crystallography. The structure of the U4 core particle shows seven proteins arranged in a ring around tightly coiled segment of RNA. ‘This structure is energetically unfavourable, stabilised only by magnesium ions that neutralise the negatively charged RNA phosphates,’ explains Oubridge. ‘It is not surprising that it took us so long to obtain enough good crystals and solve the structure to atomic resolution.’ The seven proteins each contain the Sm motif and combine to form a continuous, back-and-forth beta sheet. This ring is found in all the snRNPs except U6, which has a similar structure named LSm. The intact U1 snRNP particle has a longer RNA, the seven common Sm proteins and three others; solving it by x-ray crystallography took well over 10 years.
The technique of the future
It is unlikely that the structure of the whole complex, however, with its five snRNPs and associated proteins bound to the nucleic acid, would ever have been solved using those two techniques alone. To obtain a structure for a molecular complex as large, dynamic and fragile as the active spliceosome, only one technique will do: electron microscopy (EM), the ‘brash upstart in structural biology’, as Eric Hand, European news editor of Science, described it.
Electron microscopy as a potential tool in the structural biologist’s arsenal has been around for decades: Klug’s 1982 Nobel citation includes a reference to his ‘development of crystallographic electron microscopy’. But before the 2010s the resolution of molecular images obtained by EM remained very poor. Crystallographers and NMR spectroscopists, proud of their own ability to discern reaction mechanisms at ångstrom level, would sometimes refer to it disparagingly as ‘blobology’. Improvements in microscope power, experimental techniques and computational analysis in the last decade, however, have changed the discipline beyond recognition. Hand described it as ‘challenging crystallography in resolution and surpassing it in purview’. And while the number of x-ray structures entering the Protein Data Bank peaked in 2018, the much smaller number of EM structures is growing exponentially. ‘X-ray crystallography won’t die out completely, as it is by far the best technique for, for example, studying enzyme mechanisms in intimate detail, but electron microscopy could well supersede it for many applications,’ suggests Shaoxia Chen, who looks after the LMB’s three gargantuan Krios electron microscopes.
Experiments can only be done in vitro because if you halt splicing in cells, they die
Although it is not necessary to make crystals of a protein in order to put it under an electron microscope – which gives this technique one of its advantages over crystallography – sample preparation can still be tricky. Understanding the molecular mechanism of a dynamic structure like the spliceosome needs multiple structures, each frozen in its conformation at a different point. Clément Charenton, a postdoc in the Nagai group, explains how he and his colleagues trapped spliceosomes at different points in time. ‘These experiments can only be done in vitro because if you halt the splicing mechanism in cells, they die,’ he says. ‘It is, however, possible to set up a splicing reaction with pre-mRNA and spliceosome components in a test tube and the pre-mRNA can be mutated so the splicing process stops, yielding a sample containing complexes at exactly the same point in the mechanism.’
This general technique has so far yielded structures of human or yeast spliceosome complexes at eight different points during splicing, illustrating just how extraordinarily complicated it is. Whereas most enzyme active sites are already formed when the substrate binds, the spliceosome must assemble its active site on the nucleic acid. It must then bring the reactive groups on the RNA close together, catalyse two separate reactions with further conformational changes between them, and finally dissociate to release the products. The many complexes solved by Nagai and his co-workers, as well as the groups of Yigong Shi (Tsinghua University in Beijing, China), Reinhard Lührmann (Max Planck Institute for Biophysical Chemistry in Göttingen, Germany) and Rui Zhao (University of Colorado in the US), have enabled them to reconstruct this completely. Max Wilkinson, a graduate student in the Nagai group, has made movies of the splicing mechanism using PyMol, a molecular graphics program. Viewed in their entirety, they resemble an exquisitely choreographed ballet. ‘Some features of the complexes are easier to see and understand using these movies than from the static images alone,’ says Wilkinson.
Chemically, the reactions that are catalysed by the spliceosome are both transesterifications, in which one ester is changed into another through exchange of an alkoxy group with an alcohol. This relatively simple reaction, which requires metal ions bound to the active site, is used firstly to free the end of the 5′ exon and then, after a series of conformational changes, to join it with the 3′ exon, releasing a lasso-shaped intron known as a lariat.
Mistakes matter
It is not surprising that mistakes can occur in a process as complicated as splicing. Various forms of mis-splicing have been implicated in a growing number of diseases, and splicing defects are often seen in cancer cells. The spliceosome is, therefore, emerging as an important new drug target. Several compounds, including the macrocycle pladienolide B, have been identified as binding to spliceosome components in cancer cells and inducing cell death through splicing inhibition. It is not impossible that a compound with a similar mechanism might become a useful chemotherapy drug. Furthermore, antisense oligonucleotides that modulate splicing through binding to specific RNAs are under development for treating degenerative diseases such as amyotrophic lateral sclerosis, also known as motor neurone disease, and Duchenne muscular dystrophy.
Spinal muscular atrophy (SMA) is another degenerative disease arising from defects in the splicing process, although this time more indirectly. It is caused by the death of motor neurons, which leads to greater or lesser degrees of progressive muscle wasting. Babies born with the most serious variant will live for a few weeks or months only, whereas an individual with the mildest form can have a normal lifespan, with weakness increasing from early adulthood. Painstaking work in the early 1990s, when gene mapping was still in its infancy, tracked the cause of this disease down to defects in a gene later named SMN (for survival of motor neurons’). At about the same time, Gideon Dreyfuss of the University of Pennsylvania in the US discovered a protein that seemed to bind to many RNA-binding proteins in a unique way, and, eventually, recognised it as SMN’s protein product. ‘We found SMN to be the core of the molecular machine that constructs snRNPs, recruiting the seven proteins that form the ring structure around each snRNA,’ says Dreyfuss. ‘Cells with no SMN cannot survive at all, and its reduction leads to defective splicing and to the different types of the disease’.
Structures of the SMN protein and other components of the complex, many obtained by Dreyfuss’ group, show how it works to assemble the protein components of the snRNPs around the nucleic acid. ‘We have deduced the structures of snRNP precursors that had been predicted for decades but not seen before and revealed a link between reactive oxygen species and loss of SMN function,’ he says. ‘And we are now developing fast, sensitive assays for compounds that modulate this function, and that might prove to be useful drug leads.’
It seems likely that as more is learned about how human mRNA is synthesised and decoded, and the structure and chemistry of the molecular machines involved, further devastating diseases will become amenable to drug intervention. If so, future patients will have much to thank structural biologists like Dreyfuss and Nagai for.
Clare Sansom is a science writer based in Cambridge, UK









No comments yet