Very short delays have been measured between forward and backward photoelectrons emitted from a chiral molecule
An international research team has been able to precisely detect when electrons are ejected from a chiral molecule using an attosecond photoelectron interferometry technique. The method is so accurate that it could be applied for benchmarking quantum theories of molecular ionisation or adapted to study ultrafast processes in superconducting chiral nanotubes or spintronics devices.
When light hits a molecule, it can ionise it, causing the near-instantaneous emission of electrons. Since electrons are so light, their response was thought to be too fast to be measured experimentally, but recent developments in attosecond science have made such studies possible.
A team led by Yann Mairesse from the Centre for Intense Lasers and Applications (CELIA) in France studied the ionisation of randomly oriented camphor molecules using a special attosecond setup. ‘Our experiment revealed a very small temporal shift between forward and backward electrons —only 7 attoseconds,’ says Mairesse. ’This delay reverses if you switch the enantiomer.’
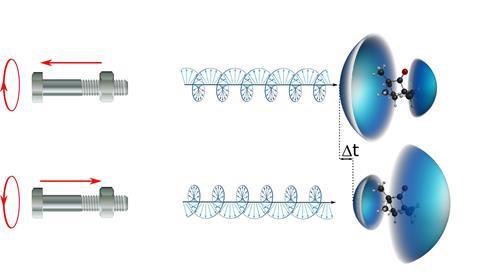
It is well known that when a chiral molecule is ionised by circularly polarised light, there is an imbalance between the intensity of electrons emitted in the forward and backward directions relative to the pointing of the light beam. ‘This asymmetry, studied by photoelectron circular dichroism (PECD), reaches several percent, some three orders of magnitude greater than the asymmetry typically encountered in longer established techniques,’ says Ivan Powis of the University of Nottingham, UK, who was not involved in the research. ‘This potentially confers on PECD a much greater sensitivity for distinguishing left- and right-handed molecules. However, full theoretical understanding of the PECD effect is challenging.’
‘When you rotate a bolt on a nut, the chirality of the thread transforms the rotation into a translation, forward or backward depending on the rotation direction,’ explains Mairesse. ‘The same happens when you photoionise chiral molecules with circularly polarised light. Remarkably, this happens even in a jet of randomly oriented chiral molecules. Our measurement was possible thanks to the very high stability of the laser source and of all optical elements, which enabled us to reach a temporal resolution of 1 attosecond.’
The researchers developed a technique using ionisation by two phase-locked laser fields to detect differential attosecond delays. They worked at wavelengths in the infrared (800nm) and ultraviolet (400nm) regions and combined linearly and circularly polarised femtosecond laser pulses. By independently controlling the chirality of the ionising and probe light pulses, the scientists could decouple the real photoionisation delays from measurement-induced ones, which can introduce inaccuracies. The photoelectrons were collected by a velocity map imaging spectrometer to obtain an angle-resolved photoelectron spectrum and compare the delays of electrons ejected in different directions. Powis calls the experimental work ‘extremely elegant, both in concept and execution’.
Norbert Böwering of the University of Bielefeld, Germany, says the work gives important new insights into chiral ionisation mechanisms. ‘It shows that inherent structural asymmetries of the examined molecules also lead to temporal asymmetries in the photoelectron emission process if the ionisation is carried out using light with inherent orientation,’ he says.
Powis believes that the method’s experimental complexity could limit its use in routine analytical applications, as there are easier ways to determine the handedness of molecules. But he says the new findings may underpin the development of other procedures to identify and quantify enantiomers. Mairesse agrees that the attosecond dynamics underlying PECD are, for now, of more fundamental interest. ’This is a first step towards understanding the transfers between the nuclear degrees of freedom, which define the chirality, and the electronic degrees of freedom, which define chemical reactivity,’ he says.
References
Beaulieu et al, Science, 2017, DOI: 10.1126/science.aao5624










No comments yet