Hydrogens seen crystal clear in small molecules
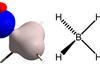
Researchers have pinned down the precise position of hydrogen atoms in crystal structures using widely accessible methods
Researchers have pinned down the precise position of hydrogen atoms in crystal structures using widely accessible methods