Mirror-symmetry breaking reaction’s mechanism solved after 25 years
By  Katrina Krämer2020-04-06T13:45:00
Katrina Krämer2020-04-06T13:45:00
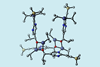
Cube escape and enzyme-like binding mechanism behind idiosyncrasies of the autocatalytic asymmetry-amplifying Soai reaction
Chemists have cracked the decades-old mystery of the mirror-symmetry breaking Soai reaction, one of the strangest and most unique transformations in organic chemistry. It turns out that an enzyme-like mechanism allows tiny chiral imbalances to create single enantiomer products from non-chiral starting materials. Understanding the Soai reaction might provide clues to how life came to be built on DNA, amino acids and sugars that exist almost exclusively in one mirror image form.