A rare modern-day sighting of a quinone intermediate
Quinones occupy a special place in my heart. And I’m not just talking about the ubiquinone cofactors in my mitochondria. Although the strange-looking quinone functional group is not immediately recognisable to many younger students of organic chemistry, it is essential to much of life on Earth and appears in several life-saving drugs. They also have a long and storied history as both intermediates and targets in total synthesis, including, least notably, in my own PhD studies.
Quinone chemistry in the laboratory shot to fame almost a century ago, when Otto Diels and Kurt Alder discovered their eponymous cycloaddition reaction using benzoquinone as the dienophile, chemistry that would later win them the Nobel prize. In the decades that followed, many landmark total syntheses exploited the use of quinone intermediates, often in Diels–Alder reactions. R B Woodward’s synthesis of reserpine, Yoshito Kishi’s synthesis of tetrodotoxin and Marshall Gates’ synthesis of morphine are just a few ultra-classic textbook examples. The strange thing is, none of these molecules actually contains a quinone! But with relatively little chemistry known, and literature searching so arduous, relying on ‘safe’ and well-precedented quinone Diels–Alder chemistry simply made sense at the time. In addition, the multiple reactive sites on a quinone (or its cycloadduct) provide a great jumping off point for further functionalisation. These days, however, it’s rare to see quinone intermediates en route to non-quinone natural products – which is to say, it’s rare to see them at all. After all, why contort a route to follow old precedent and intermediates that don’t really fit the target?
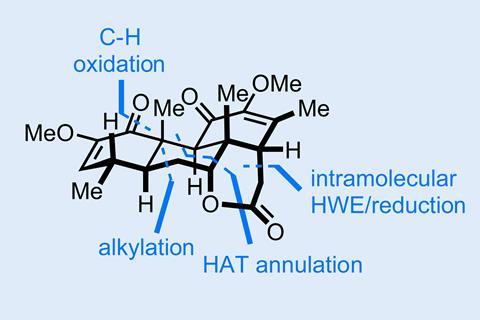
A recent example of some elegant quinone chemistry that fits a route perfectly can be found in the recent total synthesis of quassin by Sergei Pronin and co-workers at UC Irvine in California, US.1 Let’s pick things up at the start of the synthesis where the group sets the last of the molecule’s all-carbon quaternary centres.
The first step is the venerable quinone Diels–Alder reaction, but with a modern twist. Using conditions developed a few years ago by E J Corey, the team can perform the reaction asymmetrically, obtaining the cyclopentadiene adduct with near-perfect yield and enantioselectivity. The newly installed ring is then used to temporarily block the top face of the quinone, allowing the team to stereoselectively epoxidise on the convex face before it is removed in a retro-Diels–Alder at high temperature. This clever stereochemical relay allows the team to formally accomplish the catalytic enantioselective mono-epoxidation of benzoquinone – a daunting and unknown reaction. Next, the stereochemical baton is passed one more time as the newly minted epoxide is used to direct the key step in the synthesis, an iron-mediated radical annulation with a g,d-unsubstituted aldehyde. First, hydrogen atom transfer across the aldehyde’s 1,1-substituted alkene gives a tertiary radical that undergoes a Giese-type addition to the quinone, followed by enolate cyclisation onto the aldehyde (figure 1).

This is a really neat and expeditious way to unite the two fragments under very mild conditions – and with surprising stereocontrol in the initial radical addition. However, the catch is that under these conditions the final aldol closure results in the undesired stereochemistry being obtained at the alcohol stereocentre. Fortunately, the team realised that this undesired epimer was not the thermodynamic aldol product and through careful screening was able to find basic conditions under which it could be epimerised and trapped with a phosphonoacetate group (figure 2).

From here, intramolecular Horner–Wadsworth–Emmons cyclisation completes the lactone and the team is just a few functional group manipulations away from the target. Great work!
References
1 W P Thomas and S V Pronin, J. Am. Chem. Soc., 2022, DOI: 10.1021/jacs.1c12283








No comments yet