Lyconadin A
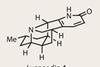
Having spent a few years working in the pharmaceutical industry, one thing I’ve learnt is that Alzheimer’s disease isn’t an easy nut to crack. Any opening into its prevention or treatment is understandably leapt upon, especially natural products such as the Lycopodium family that show activity against the disease. Members ...