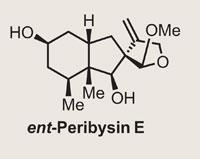
Peribysin E
Peribysin E
Many a distinguished synthetic chemist has developed a name for a particular type of natural product, often after creating a methodology well suited to making them. But Samuel Danishefsky of Columbia University (and the Sloan-Kettering Institute for cancer research) in New York City, US, has instead focused on function rather than family. His many synthetic conquests are unified by their cancer-busting potential.
The latest to succumb to his endeavours is peribysin E, a target with an intriguing mode of action. Rather than killing tumourous cells, it attacks their cell-adhesion properties, which Danishefsky suggests could make for a less toxic therapy. Inspired by this knowledge, the group went straight to the terpenes cupboard, picking up S -carvone, and then the dienes box - which I imagine is quite extensive in the Danishefsky group - before adding a little Lewis acid for a Diels-Alder very reminiscent of that seen in last month’s instalment on platencin (Chemistry World , September 2008, p38). This quickly and efficiently completed a second ring, and generated two further stereocentres. A little oxidation of the immediate product with palladium acetate gave the desired enone (figure 1).
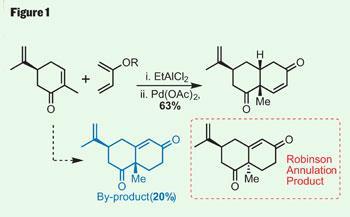
However, this reaction also turned up an intriguing enone isomer, formed by allowing warming between the cyclisation and oxidation steps. In a slight diversion from the total synthesis, the team found they could improve the yield of this by-product by tuning the reaction conditions. This might seem counter-intuitive - after all, this doesn’t help their synthetic efforts towards peribysin E - but the by-product bears a strong resemblance to product scaffold produced from a Robinson annulation. This classical reaction has been used in countless syntheses over its long history - so Danishefsky’s accidental discovery of a simple method for making the other diastereoisomer could prove very useful.
Back on track
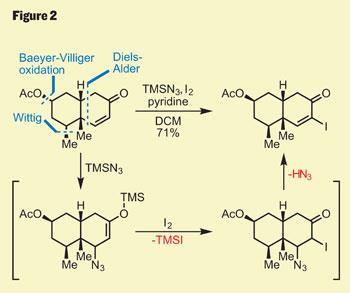
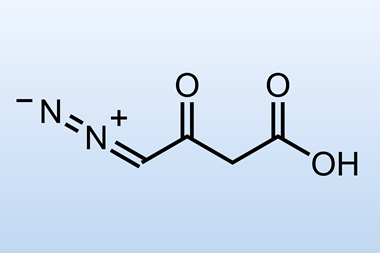
Interesting as this was, it wasn’t pushing forward their total synthesis, so those flasks went back in the freezer. Several functional group interconversions were required to elaborate their decalin core, but it was the last that caught my attention - the transformation of the enone into a vinyl iodide (figure 2). Although this reaction only involves replacement of a hydrogen atom with iodine, the method used was, to my eye, extremely elegant. Using trimethylsilyl (TMS) azide activated the enone, and was followed by conjugate addition of azide. A bit of iodine then attacked the silyl enol ether, and the unstable azide eliminated to give the desired product. I find this kind of classical chemistry inexplicably satisfying!
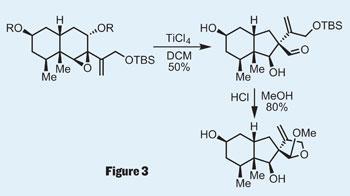
The vinyl iodide was perfectly set up for a Suzuki coupling to add the remaining carbon atoms required in the natural product. Epoxidation then led the team to the substrate required for an ambitious rearrangement reaction. In fact, the group had to try quite a few - Favorskii, benzylic acid, or semipinacol rearrangement, anyone? But the latter did the job - after a little tweaking - giving an overall ring contraction to provide the final intermediate in the synthesis; a little acid gave them the natural product (figure 3).
Or to be more accurate, it gave them the enantiomer of the natural product, as it would appear that the original assignment of the absolute stereochemistry of the peribysin E was wrong. Of course, the NMR spectra of both enantiomers are identical, with only the optical rotation giving the game away. The group were probably extremely confident of their reassignment, but just to be sure, they went through the entire route again, starting with the opposite configuration of carvone (the R enantiomer in this case). This resulted in data entirely consistent with the target, and a very nice synthesis.
Paul Docherty is a medicinal chemist based in London, UK
References
A R Angeles, S P Waters and S J Danishefsky, J. Am. Chem. Soc. , 2008, DOI: 10.1021/ja8048207










No comments yet