Exploring nickel reactivity in C–H activation chemistry
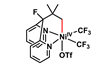
Coming closer to an alternative for palladium
Nickel has historically sat in the shadow of its ‘big brother’ palladium, though in recent years its distinct reactivity has led to it stealing some of the limelight (almost literally) with impressive applications in photoredox catalysis and reductive cross-coupling chemistry. In contrast, palladium still reigns supreme for C–H activation chemistry, with nickel being plagued by the need for extreme temperatures (>130°C) and poor functional group tolerance.