By Chris Nawrat2015-04-27T00:00:00
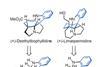
Desymmetrisation offers a neat way to add tricky features, says BRSM