By Karl Collins2014-07-03T00:00:00
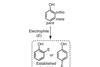
Karl Collins examines a stealthy strategy for directed C–H activation