Amide library created at speed with machine learning and stopped-flow chemistry
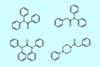
Approach used 90% less starting material than continuous flow equivalent
A team of scientists based in Sweden and the UK has developed a synthetic screening method that uses stopped-flow chemistry and machine learning to accelerate drug discovery through diversity-oriented synthesis.