Stereoselective radical coupling to rival classic Strecker synthesis for easy access to unusual amino acids
The 168-year-old Strecker amino acid synthesis might have to pack its bags to make room for a radical reaction that can make even the most exotic α-amino acids. The method has already been put through its paces in medicinal chemistry labs at pharma companies Pfizer and Bristol-Myers Squibb.
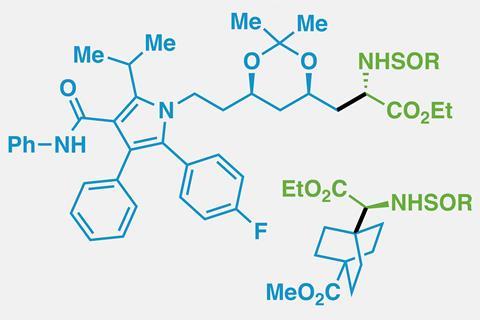
Although there are only around 20 protein-forming α-amino acids, attaching other side chains to an amino acid core gives chemists an infinite number of compounds to play with. Unnatural amino acids can be incorporated into tailored peptides, used as molecular probes to understand biological functions or designed into drugs that disrupt protein–protein interactions.
In 1850, German chemist Adolph Strecker started making amino acids by converting an aldehyde into an amino nitrile and then hydrolysing it. Over the years, the reaction has received a number of updates – there are now catalytic and asymmetric versions – but it has retained some of its drawbacks: it remains incompatible with certain functional groups and often still uses toxic cyanide reagents.
Prompted by medicinal chemists at Pfizer, whose efforts to make non-natural amino acids were thwarted by existing methods’ shortcomings, Phil Baran from the Scripps Research Institute in the US and his team came up with an entirely new way to make them. ‘It would be very hard to find a simpler way to put them together,’ Baran says.
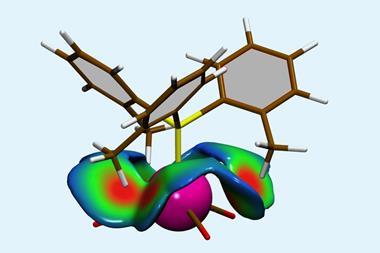
The team’s method first converts carboxylic acids – ‘the most useful and ubiquitous building block on the planet’ according to Baran – into redox active tetrachlorophthalimide esters. Then, with help from a nickel catalyst, the ester is paired with a sulfinimine as the unit carrying the amine part. Since sulfinimines are chiral around the sulfur atom, using them as single enantiomers makes the reaction asymmetric.
Baran’s team tested the reaction on more than 85 compounds, in some cases – such as for a cyclopropane amino acid – cutting the number of steps from seven to just one. It is even compatible with more outlandish side chains containing propellanes or atorvastatin.
‘It’s an incredibly useful tool,’ says Karl Collins, a medicinal chemist at Bayer Pharma in Germany and regular contributor to Chemistry World’s ‘Organic matter’ column. ‘It’s definitely something that we would also use in our lab as a simple way to make building blocks that you just can’t make otherwise.’ Four medicinal chemistry teams trialled the reaction, using it to build up amino acid libraries. Out of 30 reactions, 25 produced at least 1mg of product – enough for bioactivity testing.
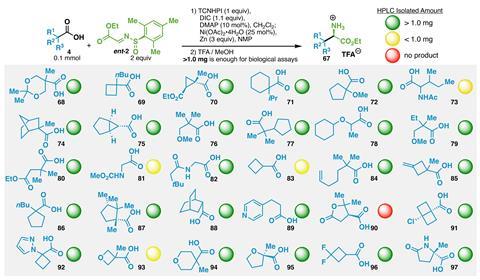
Although using a redox activator as well as a chiral auxiliary might seem wasteful, ‘from a medicinal chemistry perspective, the scale we’re working on is small enough that none of those aspects would be limiting’, says Collins. Baran adds that the auxiliary also means the diastereoisomers can be separated easily in cases where the stereoselectivity isn’t perfect.
Benzoic acid was one of a few building blocks that proved resistant to the reaction. As a workaround, Baran’s team started from benzyl bromides instead. ‘The next big step – and this is very challenging from a mechanistic perspective – is to do the reduction of aryl carboxylic acids,’ says Collins.
References
S Ni et al, Angew. Chem. Int. Ed., 2018, DOI: 10.1002/anie.201809310








No comments yet