
Up to 20% of chemical structures may be misassigned. A team of researchers has used computational NMR prediction to correct some, but other revisions remain contested
Structural and mechanistic revisions using machine learning-augmented computational NMR have caused controversy between researchers. A team analysed and corrected various unusual structures and mechanisms from the chemical literature but has since been challenged on the accuracy of their proposals. These disputes show that chemists need to strike a balance between relying on computation, experiment and intuition.
Nuclear magnetic resonance (NMR) is one of the most informative tools for organic structure determination. However, interpreting spectra is subjective and a number of molecular features can complicate this further. Quaternary carbons, for example, do not provide 1H NMR signals, meaning that in some cases large parts of a molecule become essentially invisible. Heavier atoms such as bromine and iodine can also affect chemical shifts in unusual ways. ‘Because they’ve got lots of electrons, they induce what are called relativistic effects on the chemical shift,’ explains Craig Butts, a structural and mechanistic chemist at the University of Bristol, UK. ‘This means that the typical organic chemist’s way of interpreting the spectrum starts to fall apart when there are large halogen atoms involved.’
Computational NMR methods already exist but their practicality for organic chemists is limited. Simple predictive tools available with chemical software packages often have poor accuracy. Conversely, analysis at quantum level is also possible but the technical complexity and long computation times make this unsuitable for routine use.
Andrei Kutateladze, from the University of Denver, US, and his team have developed a machine learning-augmented computational NMR method called DU8ML. It combines the speed of machine learning with the accuracy of density functional theory calculations. The system currently has a training set of 13,000 rigorously validated structures, which enables the machine learning component to recognise a wide range of functional groups.
‘We aim to identify the most challenging structural elements that often lead to misassignments in natural products,’ says Kutateladze. ‘We flag any suspect structures and double check them with the DU8ML method. From experience, we would estimate that between 12 and 20% of published structures may include a structural misassignment.’ These misassignments usually come down to stereochemistry or regiochemistry, which can be particularly problematic to assign in compounds with many quaternary or halogenated carbons.
As part of the ongoing training project for DU8ML, the Kutateladze group reports any interesting discrepancies between experiment and computation, proposing alternative structures and mechanisms to better fit the data. However, the revision process isn’t always straightforward and dialogue between researchers is often essential to arrive at the correct answer.
In 2017, Kutateladze used an early DU8ML version to analyse over 90 sesquiterpene structures, including the plant product dichrocephone A. But a year after the team had revised the compound’s hydroxyl group configuration, a total synthesis revealed that both the original and the revision were, in fact, incorrect. The additional experimental and computational data helped Kutateladze improve DU8ML. ‘It’s never an enjoyable experience when someone corrects you, but that’s how science moves forward,’ says Kutateladze. ‘Now our method performs well enough to differentiate between the dichrocephorone structures.’
Kutateladze stresses that correction is not intended as an attack on anyone’s work or professional reputation, though this is not an opinion shared by everyone.
Contested chemistry
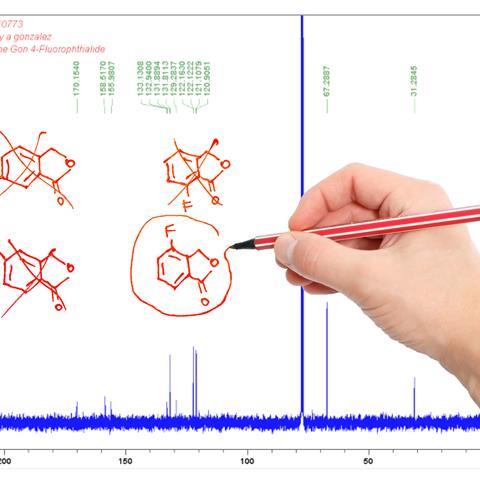
In 2008, organic chemist Metin Balci from Middle East Technical University in Turkey reported a series of bromination reactions on norbornene-type structures, suggesting a mechanism for the formation of each. The tribromide products had deceptively simple NMR spectra, which made determining their stereochemistry a stern challenge. Balci and his team performed various two-dimensional NMR experiments and ultimately used long-range coupling constants to assign the bromide groups’ relative orientations.
However, Kutateladze and colleagues proposed an alternative structure and formation mechanism for one of Balci’s compounds. ‘We are victimised,’ said Balci. ‘They are trying to refute a structure that has been proven by NMR with theory.’ DU8ML analysis indicated that the reported endo structure was a poor match for the NMR data and that exo orientation of the bromide groups was more likely. The two structures were indistinguishable with the NMR experiments performed by Balci, who had depended on the absence of long-range proton–proton coupling for his assignments.

Piqued by this criticism, Balci resynthesised the compound in question. Further NMR experiments supported his original structure, and this was confirmed by x-ray crystallography. ‘I’m curious to know what went wrong with our revision,’ says Kutateladze. ‘It is possible that we are not accounting for some interaction between the proximal bromine atoms, which the “machine” has not seen yet and therefore is not trained to correct for. Alternatively, it is possible that there is a typo in the original supporting information which fortuitously biased our conclusions in favour of the revised structure. We have asked [Balci] for a copy of the original 13C NMR spectrum to settle this issue.’
The exact cause of this discrepancy between computation and experiment has still not been determined. But for Butts, this is exactly what science is all about. ‘This whole story is about people recognising the limitations of what they have done,’ he says. ‘Balci did not do a full analysis to prove his structure incontrovertibly at the time – and that’s not unreasonable because you can’t run every experiment in the world. But then Kutateladze found evidence to support alternative answers but didn’t measure the experimental data. Two scientists just approached a very difficult problem using different methods and came to different conclusions.’
While this is not the first time that structural revisions have caused contention, the vast majority of Kutateladze’s proposals are readily accepted. In 2016, his team revised the structure of meridane, a plant natural product isolated by Pablo Chacón Morales from the University of Los Andes in Venezuela. The lack of proton signals in much of this compound had made assigning the structure particularly challenging. The following year, Chacón Morales invited Kutateladze to work together on a related natural product project.
With computation times for molecules the size of strychnine now at under 20 minutes, DU8ML already operates on a similar time scale to experimental NMR. By continuing to train the system on new and more challenging structures, the team hopes to increase prediction speed and accuracy further. ‘The ultimate goal is a broad adoption of computational methods like ours by the community,’ says Kutateladze. ‘It’s high time for everyone to augment their structural elucidation tool chest with computational NMR. Not as a panacea, but as another powerful tool in a rather extensive collection of tools for structural chemistry.’
References
I M Novitskiy et al, J. Org. Chem. 2022, 87, 13, 8589–8598 (DOI: 10.1021/acs.joc.2c00749)










No comments yet