Peptides, proteins and lipids that can enter cells and take useful payloads with them are moving towards maturity, finds Andy Extance
Without adequate blood supply, suffocating neurons die so fast that it’s no surprise that people once referred to the resulting medical condition as ‘the stroke of God’s hand’. In a stroke, neurons are deprived of oxygen, so they can’t make the energy currency molecule ATP that allows them to control the flow of ions like calcium. Consequently, the cells flood with calcium, which triggers the release of the neurotransmitter NMDA. That in turn opens ion channels that let even more calcium in, driving our neurons to produce molecules that will end up killing them.
‘In an average ischaemic stroke you lose about two million neurons a minute,’ explains David Garman, chief technology officer at biotherapeutics company NoNO in Toronto, Canada. That is the motivation for NoNO’s founders, neuroscientist Michael Salter and neurosurgeon Michael Tymianski, both from the University of Toronto, who have long sought to block the ‘ischaemic cascade’. In 1999, Tymianski’s team studied the connection in the neuron between receptor proteins that detect NMDA and a key ‘switchboard’ molecule, known as postsynaptic density protein 95 (PSD-95).1 By blocking PSD-95 production in lab neuron cultures, they cut the connection, preventing calcium-triggered damage.
Stopping human neurons making such important proteins just in case we have a stroke would have serious side-effects, so Salter and Tymianski’s teams looked for an alternative approach. But what they settled on – using peptide molecules that look like NMDA receptors to disconnect the real receptors from PSD-95 after a stroke has started – was dauntingly difficult. The peptides would have to cross two almost impenetrable obstacles – the blood–brain barrier, which keeps pathogens like bacteria out of our brains, and neurons’ own outer membranes.
Therefore the Toronto teams emulated one of humanity’s most insidious biological enemies. HIV, they knew, forces its way into cells with the help of a protein called Tat. They therefore gave chemically synthesised peptides, part NMDA receptor, part small and non-infectious Tat fragment, to rats. In 2002, they showed this approach renders rat neurons resistant to ischaemic stroke.2 ‘The company NoNO was founded to translate these findings to a means to treat humans,’ Garman explains.
Stories like NoNO’s brought high hopes for cell penetrating peptides (CPPs) like Tat. Yet 14 years later, although NoNO is now conducting phase III clinical trials on its drug, NA-1, expectations have moderated as some other efforts to exploit CPPs have foundered. In the meantime, however, scientists have been busy. Thanks to their efforts, questions about how CPPs work are being answered, and the knowledge used to add new delivery approaches to the drug development and biotechnology toolkit.
Ghosting through fatty walls
As the Canadians uncovered PSD-95, Cristina Cardoso from the Technical University of Darmstadt in Germany was wondering whether CPPs could help ‘reprogram’ mature cells. ‘We wanted to bring them back to a less differentiated state in order to multiply them and then redifferentiate them again,’ she recalls. That ability would enable growth of replacement tissue from a person’s own cells, for example, without using genetic engineering.
Cardoso was entering a field becoming riven by disagreement over how peptides penetrate cells. For example, scientists initially thought that Tat enters cells through endocytosis. In this common process, cells absorb biochemicals from their surroundings, and typically store them in endosomes and other vesicles, not necessarily making them immediately available to the whole cell. Initially working with a herpes simplex virus cell-permeable protein, VP22, Cardoso’s team added fluorescent protein labels that illuminated something surprising under their microscopes.3 ‘We could always see direct uptake, passing through the cell membrane, without needing endocytosis,’ Cardoso recalls.
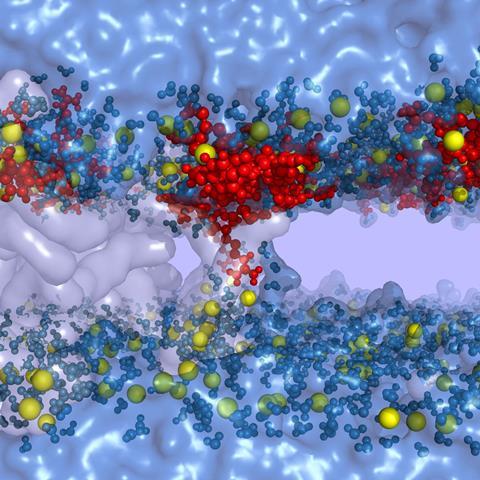
While direct uptake means peptides are more free to move after entering cells than when trapped in endosomes, the finding was controversial, Cardoso’s Darmstadt colleague Henry Herce explains. ‘Saying that something with a molecular weight of 2000 or more can go across the cell membrane is blasphemy,’ he says. This breaks ‘Lipinski’s rule of five’, an empirical set of drug discovery guidelines that suggests drugs with molecular weights over 500 are less likely to enter cells.
The German scientists have therefore studied direct uptake of Tat through both simulations and experiments.4 Protonated, positively charged, arginine amino acids in the Tat sequence bind to deprotonated, negatively charged, cell membrane fatty acids which form complex around the peptide. ‘That destabilizes the membrane,’ Herce says. ‘It forms a water-filled pore that allows other components coupled to the CPP to go from one side of the membrane to the other. Inside the cell other groups block the fatty acids, the peptide gets released, then the membrane reseals.’
Crossing the cell membrane therefore might depend on peptide structure. Cardoso, Herce and their colleagues tested this by looping peptides into cyclic structures that Cardoso says ‘we thought that for sure would kill’ the effect. Instead, they worked even better. The cyclic structures are less flexible, meaning their amino-acid side chains are aligned, speculates Cardoso, and benefit thermodynamically as they lose less entropy on binding to cells.
Delivery charges
Sónia Henriques from the University of Queensland in Brisbane, Australia, is also exploiting cyclic CPPs. She studies cyclotides, whose three disulfide bonds pull the peptide loop into a knotted coil resistant to breakdown in acidic environments and by protease enzymes in our bodies.5 The suite of methods that pierce cell membranes now includes various peptides, Henriques highlights. It also includes liposomes, fatty acid layers that surround molecules, for example RNA or DNA, and deliver them inside their targets. That’s important, because every large molecule faces specific challenges to reaching intracellular destinations, and consequently, ‘one size doesn’t fit all’, Henriques stresses.
‘You have to first think what’s the molecule you want to deliver, and what’s the target,’ she says. ‘Liposomes work very well for some molecules and tissues, but they can have low penetration and not all molecules can actually be conjugated with liposomes. Some people have created liposomes by “decorating” them with cell penetrating peptides to improve their entry into cells. And each CPP has a different internalisation mechanism dependent on peptide structure, on the cell, the cargo protein, and the concentration you are using. Some peptides prefer direct cell membrane penetration, others depend on endocytosis.’
David Liu’s team at Harvard University in the US exemplifies cargo-focus, having been inspired by pre-existing approaches to design a new method to deliver larger protein molecules into cells. In 2010, the knowledge that positively charged CPPs can enter cells gave Liu and his colleagues a hunch about how to transport proteins. ‘Perhaps superpositively charged proteins, which had a much higher net positive charge, would also be able to bind to the negatively charged outsides of mammalian cells and potently enter them,’ Liu recalls. Rather than pass through the cell membrane, they thought such large proteins would stay on cell surfaces until they were consumed through endocytosis.
Turning supernegative
Using genetically engineered cells or enzymes produced specifically via Liu’s ‘direct evolution’ approach, his team fused superpositively charged proteins to cargoes of interest.6 Their hunch was right – these proteins entered the cells efficiently through endocytosis, and Liu founded a spin-out company, Permeon Biologics, to capitalise on the findings. Sadly, Permeon’s research now has been wound down and the technology been made available for licensing or sale. Liu concedes that the superpositive proteins were slow to escape endosomes, even though his group has discovered some peptides that ‘seemed to enhance’ the process.
Yet the supercharged protein method has since surged back into the picture, thanks to a reversal of polarity. Supernegatively charged proteins look a lot like DNA, Liu explains, and can be easily complexed to positively charged cationic lipids, just as DNA is in liposomes.7 ‘Cationic lipids have been highly engineered to enter cells, escape endosomes and have other properties that make them very good for delivery,’ he says. ‘Cationic lipids will actually deliver proteins very potently into mammalian cells. In fact, it’s about 1000-fold more potent than the superpositively charged protein approach, which we suspect is in part due to more efficient endosomal escape.’
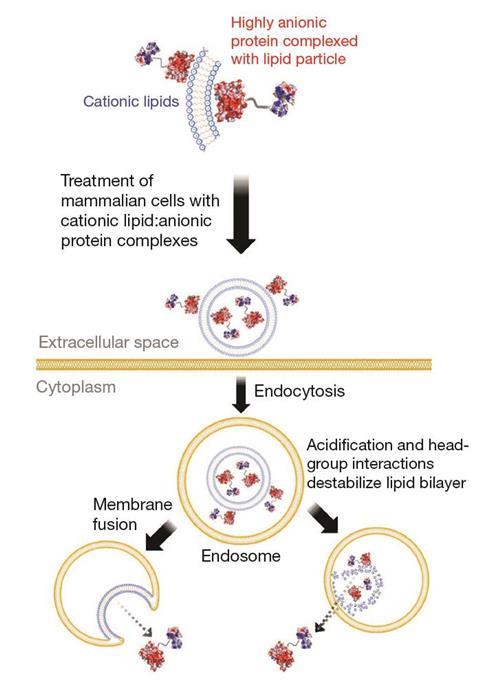
Getting large molecules into cells brings many possibilities, with Henriques wanting to use CPPs to enable the use of bacteria and algae in ‘biofactories’. ‘We can modulate intracellular properties that can then improve bacteria used to produce biofuel,’ she says. Like Herce and Cardoso, she sees this as being potentially more desirable than genetic engineering. And as NoNO has shown, getting large molecules into cells enables drugs that can do things smaller molecules cannot, she notes.
Henriques says that small-molecule kinase-inhibitor cancer drugs work well, especially in chronic myeloid leukaemia (CML), where a mutant kinase enzyme drives unregulated growth. Most famously, imatinib specifically inhibits the mutant kinase, avoiding the many other beneficial kinases in our body. However, CML patients must take such drugs constantly, and their cancers can mutate and become resistant. ‘Small molecules only target small areas in the kinase, so if there is a mutation in this protein they become inactive,’ Henriques explains. ‘Peptides can bind to a larger area of the protein target and we believe it’s more difficult to create resistance to peptides.’ Using CPP delivery also ‘opens up the options of targeting intracellular proteins that weren’t seen as druggable before’, she adds.
The Queensland scientist isn’t the first to have thought of using CPPs as kinase inhibitors. Inhibiting kinases involved in pain and cardiovascular diseases using peptide cargoes attached to Tat was among the ideas on which Stanford University’s Daria Mochly-Rosen founded spinout KAI Pharmaceuticals on in 2002. Yet after biopharma giant Amgen bought the company for $315 million in 2012 it discontinued its work on CPPs.
Xigen was also founded in 2002 in search of kinase inhibitors, as a spin-off from the University Hospital of Lausanne in Switzerland. It has developed a Tat CPP called Brimapitide (also known as XG-102 or AM-111) to carry peptides into cells, explains Jean-Marc Combette, chief executive officer of Solid Drug Development in Geneva, Switzerland, the company in charge of Xigen’s development program. The chemically synthesised peptide features d-amino acid residues, the opposite chirality to natural l-amino acids, he explains. This makes Brimapitide both resistant to breakdown by proteases.
Brimapitide remains in ‘an advanced level of clinical development in different therapeutic domains’, says Combette. In 2015, Xigen completed two phase III trials in the US to treat complaints related to inflammation in the eye, like cataracts. ‘Results from clinical studies obtained so far are being combined in order to prepare future steps required to obtain a marketing authorization,’ Combette adds.
Although many clinical trials have been conducted involving CPPs, often using cargoes attached to Tat, none has yet been approved by a medical regulator. But Henriques isn’t put off by this – she finds the fact that CPPs have made it into clinical trials ‘very promising’, and stresses the attrition rate for small molecule drugs is also high. In KAI’s case, she says, its linear CPPs would have been easily broken down in the body. By contrast, her team’s cyclotides are so robust they could even survive stomach acid – meaning they can be given in pill form.
Answering penetrating questions
Herce and Cardoso are also bringing CPP technology to drugs with industrial partners. One area they’re studying is ‘nanobodies’, aimed at targets inside cells, rather than outside like typical antibody therapies. Another is small molecules that breach the Rule of Five for reasons other than being too large, like being too fatty. However, Cardoso highlights challenges beyond designing molecules that get into cells. While making CPPs stable enough to survive stomach acid may be possible, drug makers then have to worry whether it gets from there to cells in the right part of the body.
And large molecules like Liu’s supercharged proteins bring concerns that they might trigger immune reactions when given in repeated doses. ‘In part for that reason, our lab focus is on the delivery of genome-editing proteins that in principle only need to be delivered a single time,’ he says. His team has already used its approach to deliver complexes containing Crispr-associated protein into mice to insert a gene for a fluorescent protein. ‘We’ve taken those in vivo studies quite a bit further in unpublished work,’ Liu says.
There have been over 200 failed stroke trials
Likewise NoNO only intends NA-1 to be given just once to allow patients’ bodies time to recover from stroke, avoiding stimulating immune responses with repeated doses. Garman tells Chemistry World that he’s confident his company’s evidence, spanning radiolabelling, fluorescence and mass spectrometry tests, shows NA-1 also clears other potential pitfalls and successfully accumulates in brains.
Today, in an ongoing phase III trial in Canada, paramedics give NA-1 to potential stroke patients in the ambulance on the way to the hospital. That’s because speed is of the essence, Garman says. ‘We deliver it intravenously because that’s the fastest route to get into the brain,’ he adds. NoNO is also hoping start a second phase III trial before the end of 2016.
Garman admits that he’s surprised both that development is ongoing, and that a small company like NoNO is in the unusual position of running phase III trials on its own. ‘We assumed that big pharma would have interest, but there have been over 200 failed stroke trials,’ he says. ‘It’s a very difficult field and people are risk-averse. We have to do it differently to make sure we succeed where everyone else has failed – and we’ve had to do it ourselves.’
Liu stresses how important it is for such intracellular delivery methods to be successful. ‘Delivery is an important and widespread problem facing the macromolecular sciences’ he emphasises. ‘We need as many different approaches as we can get to solve the delivery problems getting in the way of realising the research and therapeutic potential of a great many macromolecules.’
Andy Extance is a science writer based in Exeter, UK
References
1 R Sattler et al, Science, 1999, 284, 1845, (DOI: 10.1126/ science.284.5421.1845)
2 M Aarts et al, Science, 2002, 298, 846, (DOI: 10.1126/science.1072873)
3 W Derer et al, J. Mol. Med., 1999, 77, 609 (DOI: 10.1007/s001099900036)
4 H D Herce et al, J. Am. Chem. Soc., 2014, 136, 17459 (DOI: 10.1021/ja507790z)
5 Y-H Huang et al, Front. Pharmacol, 2015, 6, 17 (DOI: 10.3389/fphar.2015.00017)
6 J J Cronican et al ACS Chem. Biol., 2010, 5, 747 (DOI: 10.1021/cb1001153)
7 J A Zuris et al, Nat Biotechnol, 2015, 33, 73, (DOI: 10.1038/nbt.3081)







No comments yet